Fövárosi Szent János Kórház, II. Belosztály,1 Fövárosi Károlyi Kórház I. Belosztály és Diabetes
Szakrendelés,2 Semmelweis Egyetem, ÁOK, I. Belklinika,3 Budapest
Összefoglalás
A sulfanylurea-receptorstruktúra az utóbbi idöben került felismerésre. A receptor szerkezetének megismerésével érthetöbbé vált a sulfanylureák inzulinelválasztást serkentö hatásmechanizmusa és extrapancreaticus hatásaik eltérö érvényesülése, bár müködésének és élettani szabályozásának egyes részletei pontosan még nem tisztázottak. Az egyes sulfanylurea-vegyületek között jelentös különbség észlelhetö a szívre, az érrendszerre és a haemostasisra gyakorolt hatásukban. Ez a különbség különösen az ingerképzés és ingerületvezetés, a szívmunka és oxigénfogyasztás, a koszorú- és perifériás erek ellenállásának a befolyásolása tekintetében számottevö; nagy a jelentösége ischaemiás viszonyok között, illetve a prekondicionálás folyamata szempontjából. Ezért nem közömbös, hogy melyik sulfanylurea-készítményt alkalmazzuk szív-, ér- és haemorheologiai elváltozásokban is szenvedö cukorbetegek orális antidiabetikus kezelésekor.
Kulcsszavak: sulfanylurea-receptor,
pancreaticus és extrapancreaticus hatás, differenciált sulfanylurea-kezelés,
cardiovascularis mellékhatások
Sulphonylurea receptor structure and
its importance in the individualized sulphonylurea therapy
Summary
The structure and function of the
sulphonylurea receptor have recently been characterized. Many important details of its
physiological regulation have not yet been clarified exactly, however, analyzes of the
structural and functional properties of the receptor shed light on the insulin secretory
mechanism of sulphonylureas and their different extrapancreatic effects. There are great
differencies in the effects on the heart, the vascular system and the haemostasis among
the different sulphonyl-urea compounds. This is of particular importance from the point of
view of their influence on the arrhythmogenesis, conductivity, left ventricular function
and oxygen consumption of the heart, as well of their effect on the coronary and
peripheral blood vessel resistance. This question has a foremost importance under
pathophysiological conditions, e.g. in case of ischaemia or ischaemic preconditioning.
Therefore, the sulphonylurea drug of choice can be decisive in diabetic patients with
ischaemic heart diseases and peripheral obliterative disorders.
Keywords: sulphonylurea
receptor, pancreatic and extrapancreatic effects, individualized sulphonylurea treatment,
cardiovascular side effects
Az orális antidiabetikus kezelés irányelvei és gyakorlata a 2-es típusú
diabetesszel (2DM) kapcsolatos ismeretek gyarapodása 1,2 és a rendelkezésre álló
készítmények jelentös bövülése eredményeként 3,4,5 mára alaposan megváltozott.6,7 Elötérbe került a
nem-hypoglykaemizáló (ún. antihyperglykaemizáló) orális antidiabeticumok (OAD)
alkalmazása, mind nagyobb hangsúlyt kap a terápia fokozatos bevezetése, az algoritmus
ajánlásokban szereplö lépések következetes betartása, a sulfanylurea (SU)
-kezelésen belül pedig a differenciált kezelési stratégia. Ez utóbbi a beteg és
diabetese, illetve a különbözö SU-készítmények egyedi elönyeinek
figyelembevételével történö, a legmesszebbmenökig az individuumhoz igazított
készítményválasztás alkalmazását jelenti. 8,9
A differenciált SU-terápia mindinkább mérlegelendö szempontja a más szervek
káliumcsatornáira gyakorolt hatás, ezen belül is kiemelten a cardiovascularis hatások
alakulása.10 A SU-receptor szerkezetének és
struktúrája szervenkénti eltéréseinek megismerése e hatások megértését nagyban
segítheti. Közleményünkben a receptorstruktúrákkal kapcsolatos legújabb adatok
összegzésére teszünk kísérletet és ezen ismeretek tükrében vizsgáljuk a SU
csoportba tartozó szerek differenciált alkalmazásának egyes szempontjait,
lehetöségeit és korlátait.
A sulfanylureák inzulin-szekretagóg
hatásának mechanizmusa
Az inzulin-szekretagógok két nagy csoportra oszthatók, az ún. iniciátorokra
(ezek saját maguk képesek az inzulinelválasztás stimulálására, pl. a nutriensek
közül a glukóz, egyes gyógyszerek [pl. a SU csoport, glinidek]) és a potenciátorokra
(önmagukban nem serkentik az inzulinkibocsájtást, de fokozzák az iniciátor által
kiváltott szekréciót: pl. egyes peptidek [glukagon, glukagon-like peptid (GLP)-1],
transzmitterek [pl. az acetilkolin], aminosavak [pl. az arginin]). E különbségnek az a
magyarázata, hogy az iniciátorok képesek az ATP-érzékeny K + (KAT P) -csatorna zárására, a
potenciátorok ellenben nem.11,12 (A csatorna a nevét onnan kapta,
hogy zárt - aktív -, vagy nyitott állapota az aktuális nukleotid - elsösorban
az ATP - -tartalom függvénye. Mára tisztázódott, hogy az ATP mellett az ADP és
más nukleotidok is részt vesznek szabályozásában, amint arra a következökben még
részletesen kitérünk.)
A KAT P -csatorna nyitott vagy zárt állapota
befolyásolja a beta -sejtek membránpotenciálját. Ha a csatorna nyitva van, a K+ -ionok a csatornán keresztül a
sejt belsejéböl az extracelluláris (EC) tér felé mozognak. Ez a mozgás egy -70
mV-nyi negatív membránpotenciált tart fenn. A csatorna bezáródása a K+ -permeabilitás mérséklödéséhez
vezet, ami miatt a membrán depolarizálódik, azaz negatív töltése csökken. Ha a
nyugalmi membrán-potenciál >-35 mV, kinyílik a feszültségfüggö Ca++-csatorna, Ca++-beáramlás következik be, amely
egy enzimcascade-on keresztül a szekretoros granulumokban tárolt inzulin exocytosisát
eredményezi. A KAT P -csatorna fiziológiás regulá-torai
közé tartoznak a nukleotidok mellett a kétértékü ionok is (elsösorban a Mg ++ ).
Újabb adatok alapján az iniciátorok és potenciátorok közötti különbség
nem ennyire éles. Így pl. a GLP-1 a legújabb megfigyelések szerint önmagában is
képes a csatorna aktiválására.
A csatorna
szabályozása meglehetösen bonyolult és minden részletében ma sem tisztázott
folyamat. A felsorolt nukleotidok elvileg háromféle befolyást fejthetnek ki: gátlást,
stimulálást, illetve regenerációt. Így pl. a gátlást illetöen általánosságban
igaz, hogy az ATP mennyiségének emelkedése csökkenti, az ADP-é fokozza a csatorna
áteresztöképességét, de ionok jelenlétében a hatás módosul.13 Ha pl. az ATP Mg++-ATP formájában van jelen, hatása
gyengül. Egyes szerzök ebböl arra a következtetésre jutottak, hogy csak a nem
kötött formájú ATP tud a KAT P -csatornával interakcióba lépni.14 ADP (és egyéb nukleozid-difoszfátok)
esetében Mg++ jelenléte nélkül a csatorna
gátlódik (azaz az ATP hatásáéval egyezöen zárva van), az ion jelenlétében viszont
nyílik, söt, az ATP által zárt csatornát is nyitott állapotba viszi. Ez a
megfigyelés vezetett ahhoz a következtetéshez, hogy a csatorna szabályozásában
elsödlegesen az ATP/ADP arány, másodsorban a szabad, illetve Mg-hoz kötött nukleotid
tartalom a meghatározó. A glukóz hatására bekövetkezö inzulin-release is a glukóz
intracelluláris metabolizmusát kísérö ATP/ADP készlet megváltozásának a
következménye. A fentiek mellett a szabályozásban a cytosolban jelenlévö számos
további komponens, így a hosszú láncú acil-CoA-észterek (pl. oleoil-CoA), a
membránfoszfolipidek (pl. foszfatidil-inozitol-4,5-bi-foszfát) is részt vesznek,
csökkentve a csatorna ATP-érzékenységét. Ezzel magyarázható a nem-észterifikált
zsírsavak emelkedett szintjének inzulinszekréciót gátló hatása (pl. obesitasban,
2DM-ben). Az ionáramok tanulmányozása izolált membránrészleteken, "excised patch
clamp" technikával történik.14
A KAT P -csatorna funkcionális
károsodásának részleteit eddig három betegségben mutatták ki. Ilyen a gyermekkorban
elöforduló persistens hyperinsulinaemiás hypoglykaemia (PHHI: persistent
hyperinsulinemic hypoglycemia of infancy), amelynek hátterében a csatorna - késöbb
részletezendö - alkotóelemeinek a mutációi állnak, a MODY-2 típusú diabetes, valamint
a "maternally inherited diabetes with deafness" syndroma (MIDD). A PHHI
esetében a KAT P -csatorna a mutációtól függöen
folyamatosan zárva van, vagy a Mg++ -ADP nyitó funkciója .csökken, ami miatt a beta -sejtek
membránja tartósan depolarizált állapotú, s ez folyamatos inzulinszekréciót
eredményez. E nyitási defektussal szemben a MODY-2 és a MIDD esetében a csatorna
záródása elégtelen, ami az inzulinrelease károsodásához vezet. A MODY-2
hátterében a glukokináz enzim defektusa áll, a MIDD-ben pedig egy leucin-transzfer-RNS
mutációja miatt a mitochondrialis glukózanyagcsere károsodik, ami az ATP-képzödés
csökkenését eredményezi.15
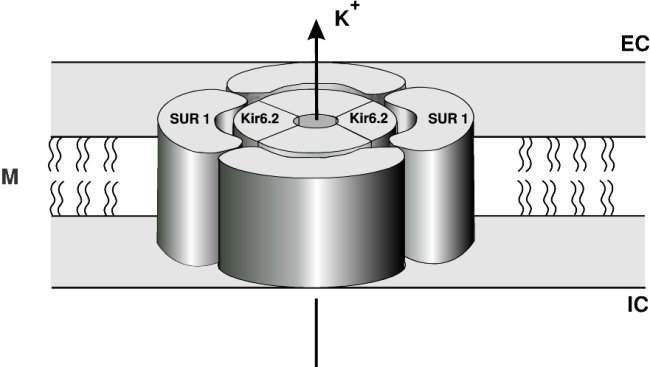
A sulfanylurea-receptor
A KAT P -csatorna két alegység négy-négy
eleméböl álló heterooktamer struktúra. Az egyik (az alfa -) alegység a
membránpórust formáló Kir (kálium inwardly rectifying=befelé irányító) család
6.X változatának valamelyik tagja (6.1 vagy 6.2), a másik, a beta -alegység a szoros
értelemben vett sul-fanylurea-receptor (SUR). A Kir tagok vannak belül, a SUR tagok
pedig, mintegy köpenyszerüen, kívül helyezkednek el (1. ábra). Általánosságban
igaz, hogy a Kir tag a K +
-áramlást, míg a SUR
tag az élettani, illetve farmakológiai ingerek, illetve hatások közvetítését
biztosítja. A Kir családon belül hét alcsoport ismert, Kir1.0-tól 7.0-ig. A KAT P -csatorna 6.X jelzésének az a
magyarázata, hogy a hatos alcsoporton belül két komponens, a 6.1 és a 6.2 is ismert,
de közülük mindenütt csak az egyik van jelen. A Kir6.1 424 (patkánykísérletböl
származó adat),16 a 6.2 pedig 390 (humán mérés)17 aminosavból áll. Valamennyi Kir
tag esetében közös, hogy két-két transzmembrán domainjük van, amelyek közül a
2-es számú tartalmazza a K + -szelektivitásért felelös három
kritikus aminosavat. Ez a Kir6.1-ben és 6.2-ben glicin-fenilalanin-glicin. Az ionáram
egyirányú voltát egy kritikus helyen lévö aszparagin reziduum biztosítja. A Kir6.1
az erek simaizomsejtjeiben és a mitochondriumok belsö membránjában van jelen, és
elsösorban a SUR 2B alegységgel asszociál. A Kir6.2 a pancreas beta -sejtjeiben, a
szív-, a sima- és a harántcsíkolt izomsejtekben mutatható ki. Legújabban neuronokban
is igazolták jelenlétét.15 Az egyes szövetekben elöforduló
alegység kombinációkat az 1. táblázatban foglaltuk össze.
A SUR és a Kir6.X gének párokban helyezkednek el az emberi genomban, egymáshoz
igen közel.14 Így pl. a szigetsejtekben lévö
receptor két alegysége, a SUR 1 és a Kir6.2 a 11-es kromoszóma rövid karján, a
11p15.1 pozícióban található. A SUR 1 gén 3' vége és a Kir6.2 5' kezdö
szekvenciája között 4900 bázispár van. A SUR 2 gén a 12-es kromoszóma rövid
karján, a 12p11.12 pozícióban, a vele társult KIR6.1 gén a 12p11.23-as pozícióban
szerepel. Itt a pontos bázispár-távolság még nem ismert. A párokban való közeli
elhelyezkedés jelentösége egyelö-re nem tisztázott. Feltehetö, hogy egy fuzionált
ösgén hasadásának és duplikálódásának a következménye. A két alegység
| SUR | Kir | A receptor farmakológiai stimulátorai |
A receptorstimuláció | ||
| alegységek | élettani szerepe | farmakológiai serkentésének lehetséges következményei |
|||
| A SU receptor | |||||
| szöveti elhelyezkedése | |||||
| Pancreas | 1 | 6.2 | diazoxid | inzulinszekréció | |
| pinacidil* | |||||
| Szívizom | 2A | 6.2 | pinacidil | ? | arrhythmogen |
| chromakalim | hatások | ||||
| nicorandil | |||||
| Vázizomzat | 2A | 6.2 | pinacidil | a fizikai terheléshez | ? |
| chromakalim | való alkalmazkodás, | ||||
| nicorandil | a fáradás mérséklése | ||||
| Simaizmok** | 2B | 6.2# | diazoxid | ? | ? |
| 6.1# | chromakalim | ||||
| pinacidil | |||||
| nicorandil | |||||
| minoxidil | |||||
| Vasculatura (artériák) | 2B | 6.1 | mint az előzőnél | értónus | hypertonia |
| izomzata | befolyásolása | ||||
| Neuron | 1 | 6.2 | mint a b-sejtnél | szinaptikus | ? |
| leírtak | transzmitter release | ||||
| Mitochondrium*** | 1?/2B ? | 6.1 | ? | ? | ? |
| *: gyengén stimulál; **: a szövetben a KATP-csatornák mellett KNDP-csatornákat is kimutattak SUR2B/Kir6.1 alegység szerkezettel; ***: a szövetekben általánosan előfordul, alegységei mitoSUR és mitoKir jelzésűek; #: a forrásokban mindkét változat előfordul | |||||
1. táblázat: A ma ismert sulfanylurea-receptorok jellemzői és szöveti típusai (a 11., 14., 15., 16., 19. irodalom alapján)
1. ábra: A KATP -csatorna sémás ábrája (a beta-sejtben elöforduló változata alapján) (Seino 15 nyomán, módosítva). M: sejtmembrán; SUR 1: a külső sulfanylurea alegység tetramerje; Kir6.2: az ionáramot közvetlenül szabályozó, belső alegység; EC/IC: extra-/intracelluláris tér; K + : az ionáram iránya
E tekintetben érdekes kivételt jelent a Kir6.2. Terminális részének
eltávolítása ugyanis nem várt módon azt eredményezi, hogy ezen alegység a SUR rész
hiányában is képes müködö ioncsatornát alkotni. Ez csak úgy lehetséges, hogy ez a
terület ún. citoplazmatikus retenciós szignál funkcióval rendelkezik, amelynek
hiányában az alegység a gardedame-mal való interakció nélkül is képes a membránba
integrálódni. Ez az önállóság egyben azt is feltételezi, hogy a Kir alegység is
rendelkezik nukleotid/SU kötö hellyel.
A SUR 1, a nagy affinitású (KD - a SU kötésre vonatkoztatott
disszociációs konstans - 2-10 nmol 15) SU-receptor 1582 aminosavból áll
(humán adat), 17 transzmembrán szegmentummal hidalja át a sejtmembránt (magyar adat 18 ). E szegmentumok domainekbe
rendezödnek, amelyek közül a TMD 0 jelü a struktúra N-terminális régiójában
helyezkedik el és öt szegmentumból áll, míg a TMD 1 és 2 jelüt egyenként hat tag
alkotja. A TMD 1 és 2 közötti területen helyezkedik el az 1-es nukleotidkötö hurok
(NBF-1: nucleotid binding fold-1), a TMD 2 után pedig, a C-terminálison, az NBF-2.
Mindkét nukleotidkötö régióban megfigyelhetö a nukleotidkötésben részt vevö
fehérjék ún. Walker-motívuma, amely
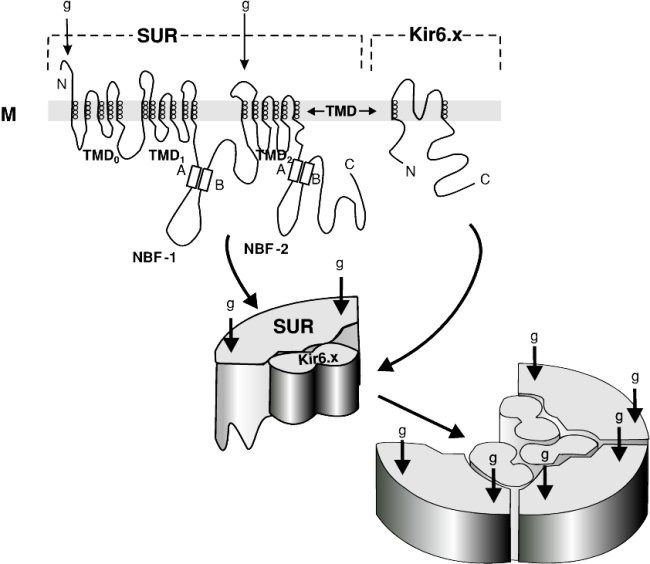
2. ábra: A SUR és Kir alegységek funkcionális kapcsolata (Babenko és mtsai [14] után, módosítva) TMD: transzmembrán domainek; NBF: nukleotid-kötö hurok; A-B: a nukleotid-kötö fehérjék ún. Walker-motívuma, az ATP-t és az ADP-t közvetlenül kötö struktúra; N illetve C: a fehérjelánc N-, illetve C-terminális vége; g: a SUR (1) alegység ez idö szerint ismert két glikozilációs helye
SUR 1 expressziót a beta-sejtek és az agyszövet mellett egyes növényekben is
megfigyeltek (ott a funkciója egyelöre nem tisztázott). Ez a receptorstruktúra
rokonságban áll a "cysticus fibrosis transmembrane conductance regulator" (CFTR), a
P-glikoprotein és a multidrogrezisztencia-asszociált fehérjékkel is. Feltételezhetö,
hogy a SU-k, vagy azok egy csoportja e rokon receptorstruktúrákkal is kapcsolatba lép,
ami szöveti (mellék)hatások létrejöttében játszhat szerepet.
A SUR 2 az
alacsony affinitású SU-receptor, amelynek SU-k iránti affinitása kb. 500-szor kisebb a
SUR 1-énél. 1545 aminosavat tartalmazó 174 kD-os fehérje (patkány adat). Szerkezete
kb. 70%-ban azonos a SUR 1-ével, a membránt áthidaló szegmentumok száma és
domainekbe rendezödése is azonos. A SUR 2 típuson belül 3 változat figyelhetö meg, a
2A, a 2B és a 2C. A 2A és B között a C-terminális 42 aminosavának összetételében
van eltérés (a SUR 2B ezen területe a SUR 1-gyel mutat hasonlóságot). A 2C pedig az
NBF-1-es régióban 35 aminosavval rövidebb a 2A-nál.
A SUR család feltételezhetö endogén
ligandja a 121 aminosavból álló endoszulfin (...(alfa- és beta -), amelyet elöször
az agyból izoláltak, késöbb azonban a szigetsejtekben, más endokrin szövetekben és
az izomzatban is kimutattak. Bizonyítást nyert, hogy a klónozott endoszulfin képes a
bevitt jelzett glibenclamidot a beta-sejt membránjáról leszorítani. Azt is igazolták,
hogy az endoszulfin önmagában, glukóz nélkül is meg tudja indítani az
inzulinelválasztást. E protein nem szekretálódik, mindvégig intracellulárisan marad,
és nagyfokú homológiát mutat egy cAMP által regulált, ismeretlen funkciójú
foszfoproteinnel. Ennek alapján feltételezhetö, hogy az inzulinelválasztás intrinsic
regulátora, esetleg az intermedier anyagcsere szignáljainak közvetítésében vesz
részt. Élettani szerepéröl ma még keveset tudunk, így az inzulinszekrécióban
betöltött esetleges szerepe sem tisztázott.
Transzgénikus egér-modellben a KATP-csatorna hiánya (a Kir6.2 gén
kiütése) károsítja mind a glukóz, mind a tolbutamid indukálta inzulinszekréciót,
de csak minimális mértékben befolyásolja a glukóztoleranciát. Az inzulin
vércukorszintet csökkentö hatása kifejezettebbé válik ezekben az állatokban. Ez
utóbbi magyarázata részleteiben nem ismert, de megerösíti, hogy az említett
csatornák a vázizmokban részt vesznek az inzulinhatás szabályozásában.15
A sulfanylurea-SU-receptor
kötödés
A KATP-csatorna élettani szabályozása
még nem ismert pontosan. Egy lehetséges modell szerint 15,19 a SUR alegység közvetíti mind az
ATP-, mind az Mg-ADP-érzékenységet. Ez a modell elképzelés azon alapul, hogy a SUR
alegységben
Az Mg-ADP egyes vizsgálati adatok szerint 11 kötödik mind a SUR, mind a Kir
alegységhez. A Kir6.2-vel való interakciója a csatorna záródásának, míg a SUR
alegységgel való kapcsolódása kinyílásának stimulátora. A csatorna aktuális
állapota e két ellentétes hatás közötti egyensúlyi állapot eredöje.
A SU-k biztosan kötödnek a receptor SUR alegységéhez. Gátolják az ide
kötödö Mg-ADP csa-tornanyitási hatását, és ennek következtében kizárólag a Kir
alegységen jelenlévö Mg-ADP csatornazáró effektusa érvényesül. Az újabb adatok
alapján az a valószínü, hogy a SU-k mind a SUR, mind a Kir alegységhez kötödnek, az
elöbbihez nagy, az utóbbihoz kis affinitással.11 Minthogy a receptor heterooktamer
szerkezetü, azaz négy
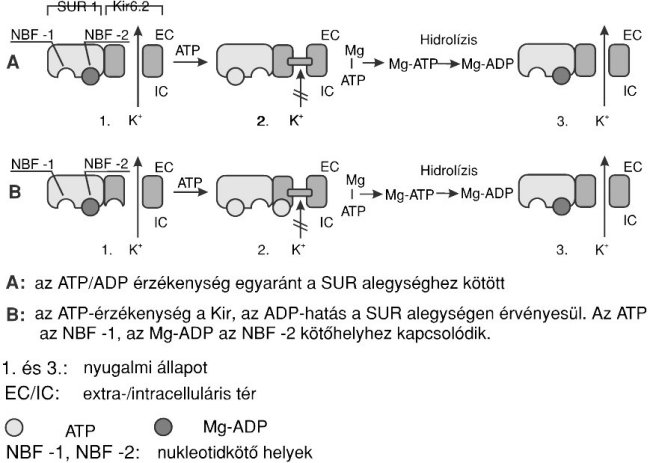
Aschroft vizsgálatai
igazolták,11 hogy a SU és rokon szerkezetü
molekulák két kémiai csoportja kapcsolódhat a SUR-k különbözö kötöhelyeivel. Az
egyik a benzamido-, a másik a szulfanil-csoport (ez utóbbit a vegyületcsoport
legismertebb, elsö képviselöje után az irodalom helyenként
"tolbutamid-csoport"-ként is említi). Csak benzamido-csoport található a
benzoesav-származékokban (ilyenek a meglitinid típusú vegyületek), csak
szulfanil-csoport szerepel a carbutamidban, a tolbutamidban, a gliclazidban, a glipizidben
és a gliquidonban. Mindkét molekulaterületet tartalmazza a glibenclamid és a
glimepirid. A benzoesav- származékokkal sztereokémiai rokonságot mutató
N-acilfenilalanin-vegyületek (pl. az A-4166 jelü nateglinid) SUR-stimuláló hatása is
feltehetöen az említett szerkezeti rokonságon alapul. A kötödés szempontjából
lényeges körülmény, hogy a SUR 1 alegység mindkét kötöhelyet tartalmazza, míg a
SUR 2A-n csak benzamido-kötöhely mutatható ki. A SUR 2A alegységü
receptorstruktúrákon tehát a benzamido-csoporttal nem rendelkezö SU-k befolyása a
Kir6.2 komponensen keresztül érvényesül.
Az említett szerzöcsoport már korábban kimutatta, hogy a tolbutamid nagy
affinitással kötödik a SUR 1 alegységhez, és így jelentösen gátolja a
pancreas-specifikus receptorstruktúrán át történö ionáramot. Kis affinitással
kötödik azonban a Kir6.2 alegységhez is. Ezzel magyarázható, hogy bár nem kötödik
a SUR 2A alegységhez, mégis megfigyelhetö kisfokú ionáramgátlás a
szívizom-specifikus receptorstruktúrán is.11 A munkacsoport azonos megfigyelésre
jutott a gliclazidot vizsgálva is: kötödése igen specifikus a SUR 1-hez (de nem
kötödik a SUR 2A-hoz), azonban a Kir6.2-höz való affinitása folytán a
szívizom-specifikus receptorstruktúrán is - csekély mértékü - ionáramgátlást
eredményez. Úgy látszik, hogy a Kir6.2-höz való gyenge kötödés a SU alapstruktúra
közös tulajdonsága.20
A SU-k receptorhoz
történö kötödésének erössége elsösorban annak függvénye, hogy a vegyület a
SUR alegységhez egy vagy két kötörégióval kapcsolódik-e (azaz jelen van-e benne
mind a benzamido-, mind a szulfanil-csoport is). A kötés erösségét azonban tovább
módosítja a kötöstruktúrák mellett jelenlévö egyéb kémiai szerkezet is. A
receptorról való leválás (a disszociáció) sebességének meghatározásában is
szerepet játszik a kötöstruktúrák száma.
Az áttekintett receptorhatások
klinikai vonatkozásai
A differenciált SU-kezelés kérdésköre jóval szélesebb az ionáramokat
befolyásoló receptorhatásoknál. Ez utóbbiak és az individuális
készítmény-választás egyes mérlegelési szempontjai, így a gyógyszerhatás
tartamát a receptorkapcsolódáson túl befolyásoló tényezök (pl. a
transzportfehérjékhez való kötödés, a szöveti és plazma felezési idö, az
elimináció helye és sebessége, aktív metabolitok keletkezése vagy hiánya), az
extrapancreaticus anyagcserehatásoknak a transzporterfunkció változásán át
érvényesülö része, valamint egyes készítmények egyedi elönyös (pl.
haemorheologiai) tulajdonságai nyilvánvalóan nincsenek egymással kapcsolatban. A
glibenclamid kizárólagos tulajdonsága, az internalizáció is független - mai
ismereteink szerint - a receptorstruktúrákkal való kölcsönhatástól. A kérdés
kiemelt jelentöségü azonban a cardiovascularis hatások szempontjából (amelyek a SU-k
esetében természetesen mellékhatások!). Feltehetöen nagy a jelentösége a
mitochondriumok KAT P -csatornáit érintö befolyás(ok)nak
is, ennek/ezek klinikai vonatkozásai azonban ez idö szerint csak kevéssé ismertek. A
ma ismert adatokat a cardiovascularis mellékhatások kapcsán az alábbiakban röviden
ismertetjük majd.
A SU-k cardiovascularis hatásai kapcsán két kérdéskört kell érintenünk. Az
egyik e vegyületek potenciális arrhythmogen tulajdonsága, a másik az ischaemiás
prekondicionálás befolyásolása.
A szívizom KATP-csatornái élettani körülmények
között zárt állapotban vannak (emiatt a SU-k csatornazáró hatása sem
érvényesülhet). Patológiás körülmények között azonban - pl. ischaemiás
stressz hatására, de feltehetöen infekciók, hyperthyreosis, toxikus noxák esetén is
- kinyílnak, s ekkor már megmutatkozhat a SU-k SUR 2A alegységgel való kifejezettebb
(és a Kir 6.2 alegységgel való gyenge) interakciója. Fontos körülmény továbbá,
hogy az interakciók következményét nagyban befolyásolják a szívizomsejtben
uralkodó metabolikus körülmények is (a nukleotidok aránya, az elözöekben felsorolt
lipid mediátorok, valamint a sejtmüködésben kulcsszerepet játszó G-proteinek
szabályozó hatása, hösokk-fehérjékkel való interakciók stb.). Ez a magyarázata az
ischaemiás szívbetegségben szenvedökön a SU-k potenciális arrhythmogen hatásának.
A SU-k többsége ugyanis ilyenkor megrövidíti a szívizomsejtek akciós
potenciáljának idötartamát, gátolja a simaizomsejtek hiperpolarizációját
(következményes elernyedését, vagyis az erek tágulékonyságát).21,22,23 A SU-k többségének ezen
kedvezötlen hatásával szemben a glibenclamid antiarrhythmiás természetünek
bizonyult: helyreállítja az ischaemiás körülmények között megrövidülö
akcióspotenciált. E hatása részben a korábban részletezett kettös
receptorkötödés (benzamido- és szulfanil-csoport), részben közelebbröl még nem
tisztázott metabolikus interakciók következménye. A kizárólagos
receptor-interakción alapuló hatás ellen szól, hogy a hasonló kötöhelyekkel
rendelkezö glimepirid szívhatásai eltérnek a glibenclamidétól: nem befolyásolja az
akcióspotenciál tartamát (de nem is rövidíti azt).
Pogátsa és munkacsoportja tisztázta, hogy a glimepirid -
eltéröen a többi SU-vegyülettöl - alig fokozza a koszorúerek ellenállását, a
bal kamra izomzatának nyomásemelkedését és legkevésbé növeli a szívizomzat
oxigénszükségletét. Megfigyelték továbbá, hogy a szívizom oxigénszükségletét
mérséklö hatását a glimepirid úgy fejti ki, hogy csökkenti a szívizomzat
összehúzódási erejét, az összehúzódás sebességét, az artériás
középnyomást, és a fentiek eredöjeként a szívmunkát. (A szívmunka
csökkentésében szerepet játszik továbbá, hogy a glimepirid, a többi SU-tól
eltéröen, nem fokozza a pancreas alfa-sejtjeiben a pozitív inotróp hatású glukagon
elválasztását.24) Leírták azt is, hogy a SUR 2A-hoz
is kötödö glimepirid és glibenclamid a SUR 1 szelektív vegyületekkel szemben
dózisfüggöen csökkentette az ischaemia/reperfusio okozta ritmuszavarokat.25 A legújabb adatok szerint nagyfokban
pancreas-szelektív SUR aktivációjú a gliclazid is, cardiovascularis mellékhatások
szempontjából ezért a második generációs SU-vegyületek többségénél
kedvezöbbnek bizonyult.20,21
A SU-k eltérö cardiovascularis hatásaiban az is közrejátszik, hogy eltérö az
affinitásuk a coronariák simaizomsejtjeiben jelenlévö KATP-csatornák különbözö
alegységeihez (SUR 2B/Kir6.2). Ezáltal hatásuk a koszorúér-átáramlásra szintén
különbözö lehet. A kérdés részleteiben egyelöre nem tisztázott.
Az új ischaemiás szindrómák (a prekondicionálás, a stunned-, maimed
myocardium és a myocardialis hibernatio) jelentösége az utóbbi idöben került a
kardiológiai érdeklödés elöterébe. A jelen munkában - a diabetológiai
irodalomban is részletesebben elemzett voltára tekintettel - az ischaemiás
prekondicionálás terápiás vonatkozásaival és a SU-készítményválasztást
befolyásoló szerepével foglalkozunk.
Igazoltnak tekinthetö a KATP-csatornák részvétele az
ischaemiás prekondicionálás mechanizmusában. E jelenség alatt azt értjük, hogy a
subletalis ischaemiás periódus(ok) védöhatású(ak) a késöbbi tartós myocardialis
ischaemia okozta szívizom-károsodás kialakulása szempontjából. E folyamat
patogenezisében két tényezö szerepe kiemelkedö, a myocardialis metabolizmus
változásáé, illetve az ischaemia indukálta reaktív hyperaemiáé. A jelen adatok
szerint e folyamatok mindegyikében mind a sarcolemmában (a szívizomsejtek
membránjában) jelenlévö ún. sarcKATP , mind a mitochondrialis membránok KATP-csatornáinak (mitoKATP ) kinyílása szerepet játszik. A
csatornák aktiválódását gátolja a megelözöen alkalmazott SU (elsösorban a
glibenclamid), de gátolja a szelektív mitoKATP-blokkoló, az 5-hidroxidekanoát
(5HD) is. A glibenclamid feltehetöen mindkét csatornát gátolja, föként az
ischaemiás prekondíció myocardialis anyagcserében bekövetkezö kedvezö hatásának
(ilyen a myocardialis ATP-tartalom nagyobb hányadának megtartása) felfüggesztésével.
Kevésbé csökkenti a postischaemiás reaktív hyperaemiát.26,27,28 Az ischaemiás prekondicionálás
folyamatában a mitoKATP-csatorna aktiválódása a fontosabb,
amit alátámaszt, hogy a mitoKATP szelektív aktivátora, a diazoxid,
experimentális körülmények között az ischaemiás prekondicionálásra emlékeztetö
védö hatást fejtett ki. Ezzel szemben a szelektív sarcKATP-blokkoló, a HMR 1883 jelü
vegyület nem befolyásolta az ischaemiás prekondicionálás protektív hatását.26
A SU-k - elsösorban a glibenclamid -, valamint az 5HD gátolják továbbá a
72 kD-os hösokk-fehérje, illetve a 4-monofoszforil lipid A (az endotoxin nem-toxikus
analógja) által létrehozott myocardiumvédelmet is.27 E folyamatok megismerése
alátámasztja azt a hosszú ideje követett klinikai gyakorlatot és az újabb kezelési
irányelvekben is megerösített ajánlást, miszerint myocardialis infarctus akut
szakában, valamint lázas infekciós betegségekben kerülendö a SU-k alkalmazása, s
- legalább átmenetileg - inzulin adására kell áttérni.
A glimepirid a glibenclamidhoz hasonlóan mindkét SUR kötöcsoporttal
rendelkezik. Ennek ellenére cardiovascularis hatásai különböznek a
glibenclamidétól, aminek magyarázata valószínüleg az, hogy kötödése a vasculatura
KATP-csatornáinak alegységeihez (SUR
2B/Kir6.1) eltérö lehet a glibenclamidétól, valamint egyéb interakciói a
membránfehérjékkel (ekto-5'-nukleotidáz, cysticus fibrosis transzmembrán regulátor
fehérje, G-proteinek, 72 kD-os hösokk-fehérjék stb.) is különbözhetnek. Egyes
adatok szerint a glimepirid az ischaemiás prekondicionálás védöhatásának
megtartása mellett 29 megnyújthatja a QT távolságot, ami
arrhythmiák felléptéhez vezethet. E hatását egy más típusú káliumcsatornára, a
HERG-re gyakorolt blokkoló hatásán keresztül fejti ki.30
Következtetések
Áttekintésünk célja az volt, hogy a KATP-csatornák szerkezetének,
müködésének, valamint a SU-kal és egyéb vegyületekkel való interakcióik
bemutatásával rávilágítsunk a differenciált SU-kezelés egyik kiemelt jelentöségü
gyakorlati kérdésének, a szívbetegségekben való alkalmazhatóságuknak a
patofiziológiai hátterére. A még sok tisztázatlan részlet ellenére elméleti
oldalról is mindinkább alátámasztottnak látszik az a klinikai megalapozottságú
álláspont,9,21 hogy a SU-k nem értékelhetök
egységes gyógyszercsoportként (legfeljebb föbb szerkezeti és hatástani
sajátosságaikban megegyezö vegyületekként), és mind nagyobb figyelmet kell
fordítanunk a készítmények egyedi elönyeinek és potenciális kockázatuknak
mérlegelésére.
Meg kell tehát fontolni, hogy azon 2-es típusú cukorbetegségben
megbetegedetteknek, akiken ischaemiás szívbetegség is fennáll, a prekondicionálás
védöhatását megtartó glimepirid, vagy a nagyfokban pancreas-szelektív gliclazid,
arrhythmia-hajlam esetén a glibenclamid, microangiopathiás károsodások esetén az
elönyös haemorheológiai sajátságú és gyökfogó tulajdonságokkal is rendelkezö
gliclazid legyen inkább a választandó SU-készítmény, ha e gyógyszercsoport
alkalmazásának javallatai és feltételei egyébként adottak. E körültekintö
mérlegeléssel (és a témánk, a sulfanylureareceptor-struktúra szempontjain kívül
esö, és emiatt nem taglalt választási szempontok figyelembevételével) lehet majd
készítmény-alkalmazásunk valóban individualizált.
Köszönetnyilvánítás
A dolgozat az ETT 03/2000 számú
tárcaszintü kutatási támogatásának keretében íródott.
IRODALOM
1. Kahn, BB: Type 2 diabetes:
when insulin secretion fails to
2. Winkler G, Cseh K: Új
készítmények és új stratégiák az
3. Owens, DR: Repaglinide -
prandial glucose regulator: a
4. Wolffenbuttel, BHR, Gomis, R,
Squatrito, S, Jones, NP,
5. Rosenstock, J, Brown, A, Fischer,
J, Jain, A, Littlejohn, T,
6. American Diabetes Association: Standards
of medical care
7. European Diabetes Policy Goup: A
desktop guide to Type
8. Baranyi É, Winkler G, Nieszner
É, Tóth J: A nem-inzulin-dependens
9. Berger, M, Mühlhauser, I,
Sawicki, P: Possible risk of
10. Nieszner É, Posa I, Kocsis E,
Pogátsa G, Préda I, Koltai M-Zs:
11. Ashcroft, FM, Gribble, FM: ATP-sensitive
K + -channels and
12. Vilsbril, T, Toft-Nielsen, MB,
Krarup, T, Madsbad, S,
13. Ashcroft, FM, Kakei, M: ATP-sensitive
K + -channels in rat
14. Babenko, AP, Aguilar-Brian, L,
Brian, J: A view of
15. Seino, S: ATP-sensitive
potassium channels: a model of
16. Inagaki, N, Tsuura, Y, Namba, N,
Masuda, K, Gonoi, T:
17. Inagaki, N, Gonoi, T, Clement,
JP, Namba, N, Inazawa, J:
18. Tusnády GE, Bakos E, Váradi A,
Sarkadi B: Membrane
19. Beech, DJ, Zhang, H, Nakao, K,
Bolton, TB: K channel
20. Gribble, FM, Aschroft, FM: Differential
sensitivity of beta-cell
21. Aschroft, FM, Reiman, F: New
insights into the molecular
22. Pogátsa G, Koltai M-Zs,
Ballagi-Pordány Gy: Influence of
23. Geisen, K, Végh Á, Krause, E,
Papp IG: Cardiovascular
24. Ballagi-Pordány Gy, Koltai
M-Zs, Arányi Zs, Pogátsa G:
25. Pogátsa G, Koltai M-Zs, Pósa
I: Vércukorcsökkentö
26. Ghosh, S, Standen, NB,
Galinanes, M: Evidence for
27. Kukreia, RC: Role of KAT P channels in heat-shock and
28. Yokota, R, Tanaka, M, Yamashaki,
K, Araki, M, Miyamae,
29. Klepzig, H, Kober, G, Matter, C,
Luus, H, Schneider, H,
30. Rosati, B, Rocchetti, M, Zada,
A, Wanke, E: Sulphonylureas