Fővárosi Szent János Kórház, III. Belgyógyászat, Kardiológiai Szakrendelés,1 II. Belgyógyászat2 és Fővárosi Károlyi Sándor Kórház, II. Belgyógyászat, Diabetes Szakrendelés,3 II. Belgyógyászat,4 Budapest
A GYULLADÁSOS FOLYAMATOK SZEREPE AZ ÉRELMESZESEDÉS KIALAKULÁSÁBAN ÉS MÓDOSULÁSUK SZÉNHIDRÁTANYAGCSERE-ZAVARBAN
Pogátsa Gábor dr.,1,3 Winkler Gábor dr.,2 Cseh Károly dr.3,4
Összefoglalás
Mind több adat támasztja alá, hogy az érelmeszesedés az érsérülés helyrehozására elinduló, de sikertelen, többlépcsős, gyulladásos folyamat végeredménye. Az érrendszert bélelő endothelium gyakran sérül a véráramlás okozta súrlódás és az elágazásokban kialakuló vérörvények hatására. A sérülések védekező gyulladásos folyamatokat indítanak el. Amennyiben a sérülés az endothelben zajló gyulladásos folyamat hatására nem gyógyul be, akkor a felhalmozódott cytokinek, illetve adhéziós molekulák további monocytákat és T-lymphocytákat tapasztanak egymáshoz és az érfal felszínére, amelyek mitogén anyagokat választanak ki. Ezek átjutnak az érfali simaizomzatba, így a gyulladásos folyamat ide is átterjed. Itt az oxidált LDL-molekulák bejutnak a monocytákba és azokat macrophagokká, majd habsejtekké alakítják. A habsejtek a subendothelialis térbe jutva felhalmozódnak és zsírcsíkokat képeznek. Amennyiben a gyógyító folyamat ekkor is sikertelen, úgy a zsírcsíkok felhalmozódása lipidmagot alkot, amelyre az érfali simaizomsejtek extracelluláris mátrix tevékenysége fibrosus sapkát von. Így alakul ki az érelmeszesedéses "plaque". A gyógyulást szolgáló gyulladásos folyamatok kialakításában részt vesz a renin-angiotensin-aldosteron rendszer is. Az angiotensin-II fokozza a cytokinek és a szöveti faktorok termelődését és hatékonyságát. Ez magyarázza az angiotensin konvertáló enzimet, illetve az angiotensin-II receptor1-et bénító vegyületek érelmeszesedést, tehát szív- és érelváltozást mérséklő hatását, amely javarészt független vérnyomáscsökkentő hatásuktól. Szénhidrátanyagcsere-zavarban felgyorsulnak a gyulladásos folyamatok, elsősorban az oxidatív stressz és a glikáció fokozódása következtében.
Kulcsszavak: gyulladás, érelmeszesedés, szív- és érszövődmények, renin-angiotensin-aldosteron rendszer
The role of inflammatory processes in the development of atherosclerosis and their modification in carbohydrate metabolic disorders
Summary
Recent data provide evidence, that subclinical, low grade inflammation could induce vascular atherosclerosis, when the inflammation proved to be ineffective in the treatment of endothelial injuries due to blood flow-induced shear stress and turbulence. Inflammation begins in the endothelial cells by the secretion of cytokines and adhesion molecules which bind monocytes and Tlymphocytes to the endothelial surface. These cells secrete mitogenic agents which could repair the injury. Inflammation propagates into vascular smooth muscle cells when these processes also proved to be insufficient to treat the endothelial injury. Oxidized LDL-molecules enter into the macrophages which developed from the monocytes and are hereby transformed to foam cells. Accumulation of foam cells produces fatty streaks in the subendothelial space. Accumulation of fatty streaks forms lipid cores when the treatment is ineffective to treat the inflammatory processes proved further to be ineffective to the treatment of the endothelial injury. Vascular smooth muscle cells generate extracellular matrix and cover the lipid core with a fibrous cap. Renin-angiotensin-aldosterone system also participates in the enhancement of these inflammatory processes. Namely, angiotensin II enhances the production and activity of cytokines and tissue factors. These explain the atherosclerosis and consequently the cardiovascular complication reducing effects of angiotensin converting enzyme or angiotensin-II1-receptor blockers, and this effect is mostly independent from their antihypertensive effect. Carbohydrate metabolic disorder enhances these inflammatory processes by increased oxidative stress and glycation.
Key words: inflammation, atherosclerosis, cardiovascular complications, renin-angiotensinaldosterone system
Bevezetés
Az elmúlt évtizedekben vált megalapozottá, hogy az érelmeszesedés érfalgyulladás eredménye. Felismeréséhez nagyban hozzájárult az abdominalisan elhelyezkedő barna zsírsejtek endokrin tevékenységének feltárása, valamint az a tény, hogy az endothelsejtek önmaguk is képesek előállítani a renin-angiotensin-aldosteron rendszer összetevőit.1 A szabad gyököknek is jelentős szerepe van a védekező gyulladásos folyamatok kiváltásában, mivel gátolják a nitrogén-monoxid (NO) hatását, kezdetben az endothel-, majd a simaizomsejtekben. 2 A téma jelentőségére utal, hogy az Amerikai Diabetes Társaság 2002. évi kongresszusán számos előadás foglalkozott a kérdéssel. Magyar sors, hogy Gerő Sándor, majd Romics László munkacsoportja3,4 már a múlt század hetvenes évei óta bizonyítják az immunológiai-gyulladásos folyamatok jelentőségét az érelmeszesedés kialakulásában, mégis csak az ezredforduló körül került a jelenség a figyelem középpontjába, Russel Ross átfogó közleményének5 megjelenését követően.
Az érelmeszesedés gyulladásos folyamat
Ép erekben az endothelsejtek által termelt nitrogén-monoxid csökkenti a vérben keringő sejtek, elsősorban a fehérvérsejtek, illetve vérlemezkék tapadási készségét az endothelsejtek felszínére,6 mérsékli az érfal áteresztőképességét, és az érfali simaizomsejtekhez diffundálva serkenti a guanilatcikláz enzim működését és ezáltal érellazulást idéz elő.7 Az endothelsejtekben képződő nitrogénmonoxid mérsékli továbbá az érfal simaizomzatának nátrium okozta fokozott érzékenységét a katecholaminok iránt, és ezáltal csökkenti a nátrium érösszehúzódást fokozó, vagyis vérnyomást emelő hatását,8 valamint gátolja az érfali simaizomsejtek szaporodását, növekedését és vándorlását.6 Az ép erek simaizomsejtjeiben termelődő NO viszont fokozza az endothelsejtek szaporodását és vándorlását, ezáltal segíti az endothelium megújulását. Érsérülés esetén pedig hozzájárulhat a gyógyulási folyamat intenzívebbé válásához.9
Ismert, hogy az érrendszert bélelő endothelréteg gyakran sérül a véráramlás súrlódása okozta nyíróerő (shear stress) és az elágazásokban kialakuló vérörvények hatására. A sérülés védekező, gyulladásos folyamatot indít el az endothelsejtekben, aminek első jele, hogy felszaporodik a gyulladást jelző, és a gyulladásos folyamatban tevékenyen részt is vevő C-reaktív protein a sérülés helyén.10 Szabad gyökök - köztük a szuperoxidanion - szabadulnak fel, amelyek gátolják az NO termelődését a nitrogénmonoxid-szintáz enzim tetrahidrobiopterin kofaktorának oxidálása, és ezáltal az enzim működésének mérséklése révén.11 Továbbá csökkentik az NO szöveti szintjét, mivel azt peroxinitrit-ionná alakítják.7 Következésképpen romlik az endothelsejtek NO-anyagcseréje. Az érsérülés fokozza az endothelsejtek endothelin és plasminogenaktivátor-inhibitor (PAI-1) termelését is. Erre megnő az endothel áteresztőképessége és adhéziós molekulák jelennek meg az endothelsejtek felszínén. Az adhéziós molekulák a vérben keringő sejteket egymáshoz (L-szelektin, integrinek, ICAM-1 stb.) és az endothelhez (E-szelektin, Pszelektin, VCAM-1 stb.) tapasztják, így monocyták, vérlemezkék és T-lymphocyták tapadnak meg az endothel felszínén.5 Ez a jelenség az úgynevezett "endothel-dysfunctio", amely az endothelsejtnek az érsérülésekre adott első válasza. E válasz eredményeképpen - az érsérülés helyrehozása érdekében - az endothelsejtek addigi antikoaguláns működése prokoaguláns működéssé változik.10
Amennyiben ez a gyulladásos válasz nem elegendő a kiváltó sérülés megszüntetésére, a gyulladásos folyamat átterjed az érfali simaizomsejtekre.5 A szabad gyökök további felszaporodása miatt, az endothelhez hasonlóan, az érfal simaizomsejtjeiben is romlik az NO-anyagcsere.9 Így a fehérvérsejtek és az azokból kiszabaduló, gyulladást serkentő cytokinek eljutnak az érfal simaizomsejtjeihez is és megindítják azok szaporodását és vándorlását.5 A cytokinek termelődésében központi szerepe van az LDL-molekuláknak. A szabad gyökök ugyanis oxidálják az LDL-molekulákat, amelyek így a "scavenger" receptorokhoz kötődve bejutnak a monocytákba, illetve a monocytákból képződött macrophagokba.12 A macrophagokba bejutott, oxidált LDL-molekulák - ellentétben a nem oxidált LDL-molekulákkal - kikerülnek a szabályozási folyamatok alól. Ezért ott nagymértékben felhalmozódnak és a macrophagokat habos sejtekké alakítják, miközben azok a subendothelialis térbe jutnak. Itt a habos sejtek felhalmozódása zsírcsíkok ("fatty streak") kialakulásához vezet. A zsírcsíkok megjelenése az érsérülés helyrehozását célzó, gyulladásos folyamatok második lépcsőjének jelzője,13 és egyúttal az érelmeszesedés korai jele is.14 A zsírcsíkokból gyulladást serkentő cytokinek szabadulnak fel, amelyek még fokozzák az érfali simaizomsejtek és a vérlemezkék összecsapódását, és az összecsapódott sejtekből erőteljes mitogén anyagok felszabadulását,15 amelyek az érelmeszesedés folyamatának továbbvivői. Az oxidált LDL-molekulák nagymértékű atherogenitását az is jelzi, hogy belőlük telítetlen zsírsavak szabadulnak fel, amelyek apoprotein-B és foszfolipid-molekulákhoz kapcsolódva erősen citotoxikus hatásúvá válnak. Ezek a citotoxikus zsírsavak serkentik az adhéziós molekuláknak (VCAM-1, ICAM-1) és a monocytákat kémiai úton vonzó fehérjéknek (MCP-1) a megjelenését, a szöveti faktorok (cytokinek, adhéziós molekulák stb.) termelődését ("expression"), a T-lymphocyták és a vérlemezkék tevékenységét, valamint az érfali simaizomsejtek növekedését. Tovább fokozzák az endothelsejtekből az endothelin, illetve a PAI-1 felszabadulását, illetve bénítják az NO (EDRF) anyagcserét.16 A HDL-molekulák mérséklik az LDL-molekulák monocytakötődést növelő hatását az endothelsejtekben.17 A oxidatív stressz, a proteinkináz-C és a glikációs végtermékek receptorainak gyulladás kiváltotta fokozott működése T-lymphocyták bevándorlását indítja el az intimába, amely ugyancsak termel gyulladást serkentő cytokineket.18 A glikációs végtermékek receptorainak megnövekedett aktivitása és az oxidatív stressz fokozódása serkenti mind a kappa-b-sejtmagfaktor (NF-kb), mind az "aktivátor protein-1" gén átírását. Ezek az anyagok - amelyek mind nagy mennyiségben találhatók az atheromában - szabályozzák elsősorban az érelmeszesedés számos tényezőjének, vagyis a gyulladást elősegítő/közvetítő anyagok (MCP-1, interleukin-1, TNF-a, LCAC, LCAM stb.) génexpresszióját és -kódolását.19 Az oxidált LDL-molekulák beépülnek az immunkomplexekbe is, ezáltal befolyásolják az immunfolyamatokat20 és megváltoztathatják a gyulladást serkentő cytokinek (IL-1, TNF-a, MCSF stb.) hatására az LDL-receptor génátírását, ami a gyulladásos folyamatok további bővüléséhez, "circulus vitiosus" kialakulásához vezet.21
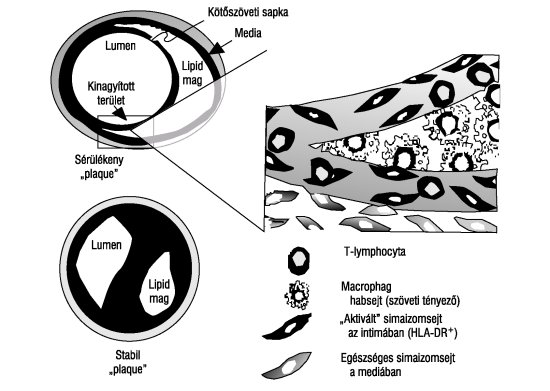
1. ábra. Sérülékeny és stabil "plaque". A habsejtek az érfali simaizomsejtekben fokozzák az extracelluláris mátrixképződését, ami a "plaque" fibrosus sapkaképződésének fontos szakasza. Az érlumenbe nem domborodó "plaque"-ok repednek meg elsősorban, mivel ezekben a simaizomsejtek száma kicsi. A lumenbe domborodó stabil "plaque" fibrosus sapkája vastag és ellenállóbb
A gyulladásos folyamatok további bővülése a sérülés helyén érfalvastagodással jár, ami a "plaque" kialakulásának kezdete. Ez a "kezdeti plaque" még nem domborodik be az érlumenbe, mert az érfal tágulása (remodelling) kiegyenlíti a lumenbeszűkülést.5 Amennyiben az érsérülés még mindig fennmarad, úgy további gyulladásos sejtek halmozódnak fel a sérülés helyén. Ezek további hidrolitikus enzimeket, cytokineket, chemokineket és növekedési faktorokat választanak ki az érsérülés helyrehozására. Amennyiben így sem sikerül a sérülés kijavítása, úgy sejtkárosodást, gócos sejtelhalást okoznak, vagyis sejtbomlás-termékeket tartalmazó lipidmaggá (lipid core) alakítják a "kezdeti plaque"-ot. Az így kialakult lipidmag fölé az érfali simaizomsejtek kollagénképzése fibrosus sapkát von, amelynek lumenbe domborodását a kompenzáló értágulat már nem tudja kivédeni. Így alakul ki a lumenbe domborodó "plaque"5 (1. ábra). Fontos felismerés, hogy az érlumenbe domborodó "plaque" vastag fibrosus sapkája ritkábban reped meg, mint a még be nem domborodó "plaque"-é. A be nem domborodó "plaque" gyakoribb megrepedésének oka, hogy az atheromán belüli immuninger - a T-sejtek interferon-gammaelválasztása révén - ebben az időszakban még gátolja a kollagénháló megerősödését.22 A fibrosus sapka gyengeségén kívül a "plaque"-on belül képződő mikroerek megrepedése is előidézhet "plaque"-repedést.22
Így a véráramlás változása is kiválthat védekező érfalgyulladást, vagy annak sikertelensége esetén érelmeszesedést. A véráramlás csökkenése ugyanis fokozza azoknak a gyulladást serkentő cytokineknek (ICAM-1, PDGF, TF stb.) a génexpresszióját, amelyeknek az úgynevezett promoter regiója érzékeny a nyíróerő csökkenésére.5
Szénhidrátanyagcsere-zavar és az érelmeszesedés gyulladásos folyamata
Nemcsak manifeszt cukorbetegségben, hanem a szénhidrát-anyagcsere azt megelőző zavaraiban, így IGT-ben is kifejezettebbé válnak az érelmeszesedés gyulladásos folyamatai. Fokozódik ugyanis az oxidatív stressz, amely módosítja számos cytokin termelődését és hatását, továbbá csökkenti a szövetekben az NO hatékonyságát. Ezáltal növeli a gyulladásos folyamatok, valamint a véralvadás intenzitását, módosítja az érszabályozást, sőt hozzájárul a hasnyálmirigy béta-sejtjeinek működésromlásához is.23 Szénhidrátanyagcsere-zavarban az oxidatív stressz fokozódásának oka egyrészt az antioxidáns rendszer hatékonyságának csökkenése,24 másrészt a vércukorszint emelkedése. A vércukorszintnek már az étkezést követő, átmeneti emelkedésére is, de tartós emelkedésekor még inkább nagymennyiségű szabad gyök képződik a glukóz autooxidációja, a poliol anyagcsereút sejten belüli fokozódása, a glikálódási folyamatok intenzívebbé válása,25 illetve a redox rendszer kiegyensúlyozatlansága26 következtében.
A glikálódás hatására az LDL-molekulák könnyebben oxidálódnak, fokozottabban bontják az NO-t, rontják az NO felszabadulását szabályozó jelátviteli rendszer működését,27 vagyis atherogenebbé válnak. Cukorbetegségben jelentősen megnő a vérben a 8-epi-PGF2a szintje, amely a ciklooxigenáz enzimúttól függetlenül, peroxidáció által képződik arachidonsavból és kifejezett érszűkítő hatása van.25 Inzulinrezisztenciában nagymértékben megnő a szabad zsírsavak felszabadulása a zsírszövetből.28 A felszabaduló szabad zsírsavak egyrészt a proteinkináz-C jelátvivő rendszeren át, másrészt az NO-szintázagonista foszfatidilinozitol-3-kináz (Pl3K) enzim gátlásán keresztül rontják az NO termelését és hatékonyságát.29
A vércukor-emelkedés fokozza mind a kappa-b-sejtmagfaktor, mind az "aktivátor protein-1" génjének átírását, ennek következtében tovább növeli az oxidatív stresszt és így tovább csökkenti az NO-szintet.30 A cukorbetegségben gyakori lipidelváltozás (szabadzsírsav- és VLDL-felszaporodás) tovább fokozza a kappa-b-sejtmagfaktor képződést és ezáltal az adhéziós molekulák és cytokinek mennyiségét.31
A szénhidrátanyagcsere-zavar az érelmeszesedést elősegítő gyulladásos folyamatok serkentése mellett közrehat a "plaque" instabillá válásában is, mivel csökkenti a "plaque" stabilitását biztosító fibrosus sapka kialakulásához és vastagodásához szükséges kollagén mennyiségét. Az endothelsejtek ugyanis olyan cytokineket választanak ki, amelyek csökkentik az érfali simaizomsejtekben a "de novo" kollagénképzést.32 A szénhidrátanyagcserezavar pedig elősegíti a kollagénállomány csökkenését a mátrix-metalloproteináz enzimek serkentése révén.33 Az így könnyebben bekövetkező "plaque"-repedés következményeinek súlyosságát még úgy is fokozza, hogy a "plaque"-ban véralvadást serkentő szöveti faktorok szaporodnak fel,34 amelyek még jobban elősegítik a megrepedt "plaque" feletti vérrögösödést, az amúgy is előnytelenné vált véralvadási állapotban.
A vércukor-emelkedés az érfali simaizomsejtekben is fokozza a proteinkináz-C, a kappa-b-sejtmagfaktor és a glikációs végtermékek receptorainak tevékenységét. Ezáltal tovább növeli az oxidatív stresszt a szabad gyökökben amúgy is gazdag érfali simaizomsejtekben, amelyeknek egyébként is lényeges szerepe van az érelmeszesedés kialakulásában. A macrophagokban gazdag zsírcsík-képződmények az érfal középső rétegébe érve fokozzák a frissen keletkezett sérülésekben az extracelluláris mátrix képződését, amely a "plaque" fibrosus sapkaképződésének fontos lépése. Azok a "plaque"-ok repednek meg elsősorban, amelyekben a simaizomsejtek száma kicsi.30 Fukumoto munkacsoportja34 cukorbetegek előrehaladott érelmeszesedéses elváltozásaiban jelentősen kevesebb simaizomsejtet észlelt, mint az anyagcsere-egészséges egyénekében. Magyarázata, hogy az érfali simaizomsejtek apoptosisáért főleg a glikált és oxidált LDL-molekulák felelősek.35
A vércukor-emelkedés a vérlemezkékben is fokozza a proteinkináz-C működését, növeli a szuperoxidanion-gyök képződését és rontja a nitrogén-monoxid hatását.36 Cukorbetegek vérlemezkéiben felborul az alakváltozásukat és thromboxanképzésüket szabályozó kalcium-homeostasis.37 Vércukor-emelkedéskor a csökkent NO- és prostacyclinképzés, valamint a fokozott fibrinogen-, thrombin- és von Willebrand-faktor-képzés következtében megváltozik a vérlemezkék fibrinkapcsolata.35 A cukorbetegségben csökkent fibrinolysis, a csökkent antithrombin-III és protein-C, illetve a fokozott faktor-VII- és PAI-1-szint növeli a megrepedt "plaque" felszínén a thrombus kialakulásának esélyét.38
2-es típusú cukorbetegségben emelkedett mind a szubklinikus lefolyású idült gyulladás fennállását, mind az alapvető immunológiai folyamatok fokozottságát jelző C-reaktív protein szintje.39 Ez is magyarázza, miért jelentkezik szénhidrátanyagcsere-zavarban korábban és súlyosabb formában az érelmeszesedés.25
Angiotensin és az érelmeszesedés gyulladásos folyamata
A cytokinek termelődését és hatását a renin-angiotensin-aldosteron rendszer is módosítja. Az angiotensin-II serkenti a NADH/NADPH-oxidáz enzim működését és ezáltal növeli az oxidatív stresszt.40 Továbbá fokozza a simaizomsejtekben a foszfolipáz-C enzim működését, emelve a sejten belüli kalciumszintet. Növeli a fehérjeszintézist, aminek következménye a simaizomhypertrophia,41 illetve a simaizomsejt-vándorlás.42 Fokozza a lipoxigenáz enzim működését, s így az LDL-molekulák oxidációját,43 valamint a gyulladást.44 Serkenti a növekedést előmozdító folyamatokat,45 illetve a monocyták aktiválódását és kötődését,46 aktiválva a vérben keringő, oldott állapotban levő gyulladásos faktorokat (C-reaktív protein, E-szelektin, MCP-1, VCAM-1 stb.)47 és befolyásolja az endothelsejtek,48 valamint a macrophagok49 működését és vándorlását.50 Az angiotensin konvertáló enzim és az angiotensin-II jelenléte az atheromában szintén alátámasztja az angiotensin peptidek szerepét az érelmeszesedés kialakulásában.51
Angiotensin konvertáló enzimet (ACE) vagy angiotensin1-receptort (ATR) gátló vegyületek hatása az érelmeszesedés gyulladásos folyamataira
Az ACE-t vagy ATR-t bénító vegyületek egyrészt úgy csökkenik az érelmeszesedés kialakulását,52 hogy gátolják az angiotensin-II-nek a szuperoxidanion képzését fokozó hatását és ezáltal javítják az endothelsejtek működését,48,53 Az ACE-gátlók csökkentik a bradikinin lebomlását, növelve az NO képződését,47 és a PAI-1 termelődését, amin keresztül fokozzák a fibrinolysist.54
Az ATR-t blokkoló losartan például csökkenti a monocyták CD11b-expresszióját, a vérlemezkék összecsapódását, az adhéziós molekulák (VCAM-1, ICAM-1) számát, valamint az E-szelektin és C-reaktív protein hatását már olyan adagban, amelyben a vérnyomást még nem befolyásolja.52 Apolipoprotein-E hiányos egerekben a losartan csökkenti az LDL-peroxidációt.12 Valószínű tehát, hogy a losartan elsősorban az adhéziós molekulák és cytokinek keletkezésének bénítása révén gátolja a zsírcsík képződést, vagyis a korai érelmeszesedés kialakulását.52 Az ACE-gátlás közvetlen szerepet játszik tehát az endothelműködés javításában és megbénítja az endothelsérülést követő intima-hyperplasia sejten belüli kulcslépéseit.55
A gyulladásos folyamat felderítésének lehetőségei
Az ezredforduló körül vált ismertté, hogy a C-reaktív protein - amely a gyulladásos folyamatok érzékeny jelzője - az ischaemiás szív- és érelváltozások önálló, független kockázati tényezője.56 A C-reaktív protein képződését a cytokinek, elsősorban az interleukin-6 indítja el a májban,57 majd nagy mennyiségben jelenik meg az érfalsérülés helyén, ahol fokozza a monocytákban, illetve az azokból képződött macrophagokban a szöveti faktorok képződését és az adhéziós molekulák kialakulását.10 Szoros összefüggés észlelhető a szervezet -- elsősorban visceralis - zsírtömege és a C-reaktív protein vérszintje között.58
Számos epidemiológiai tanulmány (Multiple Risk Factor Intervention Trial, Women's Health Study, MONICA Augsburg Cohort, Helsinki Heart Study, Caerphilly Study, British Regional Practice Study, Insulin Resistance Atherosclerosis Study ) bizonyítja, hogy a C-reaktív protein szintje megbízhatóan jelzi a szív- és érkockázat nagyságát.59 A "Women's Health Study" ezért találta érdemesnek C-reaktív protein szűréssel is kiegészíteni a lipidszűrést, elsősorban egészséges, postmenopausában levő nők szív- vagy érrendszeri kockázatának felderítésében. Az amyloid-A, interleukin-6 és oldott ICAM cytokinek vérszintemelkedése sokkal kevésbé megbízható.56 A C-reaktív protein szintjére alapozott szűrés további előnye, hogy C-reaktív protein esetében nincs napszaki vérszintingadozás, ellentétben a többi cytokin (interleukin-6 stb.) vérszintjének kifejezett napszaki eltérésével.59 A meghatározás módja egyszerű és nem költséges.
A közelmúltban felfedezett disintegrinek cysteinben gazdag és metalloproteinázt tartalmazó membránfehérjék, amelyek a cytokinek befolyásolásán keresztül a sejtek közötti kapcsolatokat szabályozzák és elősegítik a cytokinek vedlését a fehérvérsejtek felületéről. A levedlett cytokin-molekulák vérszintjéből ugyancsak következtetni lehet gyulladásos folyamat esetleges fennállására.53
Az érelmeszesedés gyulladásos folyamatának befolyásolási lehetőségei
Az aspirin előnyös védőhatása a szív- és érbetegségek kockázatának csökkentésében - a prostaglandinképződésre kifejtett hatás mellett - azon is alapul, hogy csökkenti az érelmeszesedés kialakulásában központi szerepet játszó gyulladásos folyamatokat. Erre utal a C-reaktív protein szintjének aspirin profilaxis hatására bekövetkező csökkenése, illetve e szintcsökkenés és a cardiovascularis kockázat mérséklődése között kimutatható egyenes arányú összefüggés.60 A "Women's Health Study" felveti a 3-hidroxi-3-metilglutarilkoenzim-A-reduktáz enzim bénításának előnyét is a szív- és éresemények megelőzésében, kockázatuk csökkentésében.56
A statinok gyulladáscsökkentő tulajdonságát igazolja a C-reaktív protein szintjét csökkentő hatásuk.61 A statinkezelés jelentősen csökkenti a C-reaktív protein mennyiségét a plazmában. Keringési kockázatot csökkentő hatása a magas C-reaktív protein szintű egyénekben a legkifejezettebb,62 még akkor is, ha lipidszintjeik alig emelkednek a normális szint fölé.63 A statinok kifejezetten alulszabályozzák az ATR expresszióját és hatásait.64
Az "Insulin Resistance Atherosclerosis Study" szerint szoros, egyéb tényezőktől független összefüggés áll fenn a C-reaktív protein szintje és a szöveti inzulinérzékenység nagysága között.62 Az összefüggés fennállását alátámasztja, hogy a testsúlykorrekció is csökkenti a C-reaktív protein szintjét.65 Feltétezhető tehát, hogy mind a fizikai tevékenység, mind a szövetek inzulin iránti érzékenységét fokozó (insulinsensitizer) gyógyszerek (metformin, glitazon) részben a gyulladásos folyamatok csökkentése révén mérséklik a szív- és érrendszeri kockázatot. Eldöntetlen, hogy az összefüggés közvetlenül vagy közvetve, a túltáplálás kiváltotta cytokin-túltermelés, a kockázati tényezők halmozódása, a máj akut fázis fehérjéinek szintézise, illetve a test zsírösszetételének megváltoztatása révén alakul ki.62 Anyagcsere-egészséges emberekben a plazma C-reaktív protein szintje sohasem haladja meg a 3 mg/l értéket.65
Az inzulirezisztenciát az inzulinhatás kifejeződésében kulcsszerepet játszó két sejten belüli jelátviteli út egyensúlyzavara idézi elő. E két út egyike a foszfatidilinozitol-3-kináz (Pl3K) enzimműködés, amely az inzulinfelvételt és az endothelben az NO hatását fokozza. A másik út a sejten kívüli jelszabályozó kináz enzimrendszer (ERK) mitogén által aktivált proteinkináz (MAPK) enzimjének működése, amely az inzulin növekedést kiváltó hatását közvetíti. Az ERK MAPK enzimjének működését számos hatás (pl. hyperglykaemia, angiotensin-II, IGF1, érfalfeszülés és -nyújtás stb.) serkenti, és túlsúlyba jutása a Pl3K enzimműködéssel szemben nemcsak a metabolikus szindróma, hanem az "endothel-dysfunctio" kialakulását is előidézi66 (2. ábra). Az endothelsejtek adhéziós molekuláinak működését szabályozó, gyulladást serkentő cytokinek negatív szabályozása alig ismert. Nemrég derült ki, hogy negatív szabályozásuk a magreceptorok peroxisoma proliferator activált (PPAR)-a receptorán keresztül történik, a kappa-b-sejtmagreceptor bénítása révén. Az érrendszernek szinte minden jelentősebb sejtje kifejt valamilyen működést a magreceptorok ezen csoportján keresztül. A PPAR-alfa receptorok serkentői a többszörösen telítetlen zsírsavak (pl.: dokozahexénsav, fenofibrát). Ezért is fontos a telítetlen zsírsavak és fenofibrátok alkalmazása az érelmeszesedés folyamatának mérséklésében,67 és ezért hasznos nemcsak metabolikus szindrómában, hanem "endotheldysfunctió"-ban is a glitazonok használata az érelmeszesedéshez vezető gyulladásos folyamatok mérséklésére,66 szénhidrátanyagcsere-zavart javító hatásuk mellett.
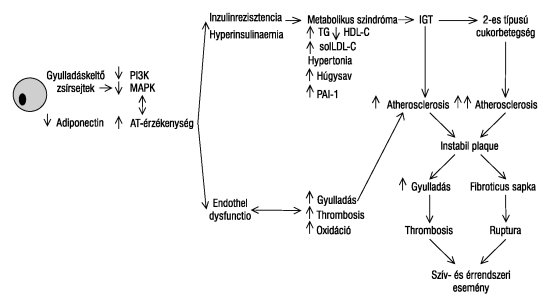
2. ábra. Gyulladáskeltő barna zsírsejtek szerepe a szív- vagy éresemények kialakulásában. Az inzulinhatás két sejten belüli jelátviteli út egyensúlyán alapul. Egyik út a foszfatidilinozitol-3-kináz (Pl3K) enzim működés, amely az inzulinfelvételt és az endothelben a nitrogén-monoxid működést fokozza. A másik út a sejten kívüli jelszabályozó kináz enzimrendszer (ERK) mitogén által aktivált proteinkináz (MAPK) enzimjének működése, amely az inzulin növekedést kiváltó hatását közvetíti
A jelenleg folyó klinikai farmakológiai vizsgálatok szerint68 a proteinkináz-C béta-módosulatát gátló vegyülettel jelentősen javítható cukorbetegségben a microalbuminuria és a diabeteszes nephropathia.
Irodalom
1. Dzau, VJ: Molecular and physiological aspects of tissue renin-angiotensin system: emphasis on cardiovascular control. J Hypertens 6 (Suppl. 3.): 7S-12S, 1988.
2. Lüscher, TF, Noll, G: Endothelium dysfunction in the coronary circulation. J Cardiovasc Pharmacol 24 (Suppl. 3.): 16S-26S, 1994.
3. Horváth M, Onody C, Gerő S: Cell-mediated and humoral immune response in various vascular diseases. Allergol Immunopathol 8: 81-86, 1980.
4. Horváth A, Füst Gy, Horváth I, Vallus G, Duba J, Harcos P, Prohaszka Z, Rajnavölgyi E, Jánoskuti L, Kovács M, Császár A, Romics L, Karádi I: Anti-cholesterol antibodies (ACHA) in patients with different atherosclerotic vascular diseases and healthy individuals. Characterization of human ACHA. Atherosclerosis 156: 185-192, 2001.
5. Ross, R: Atherosclerosis - an inflammatory disease. Review. N Engl J Med 340: 115-126, 1999.
6. Garg, UC, Hassid, A: Nitric oxide-generating vasodilators and 8-bromocyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest 83: 1774-1777, 1989.
7. Loscalzo, J: Nitric oxide and vascular disease. N Engl J Med 333: 251-253, 1995.
8. Tolins, JP, Shultz, PJ: Endogenous nitric oxide synthesis determines sensitivity to the pressor effect of salt. Kidney Int 46: 230-236, 1994.
9. Fukuo, K, Inoue, T, Morimoto, S, Nakahashi, T, Yasuda, O, Kitano, S, Sasada, R, Ogihara, T: Nitric oxide mediates cytotoxicity and basic fibroblast growth factor release in cultured vascular smooth muscle cell: a possible mechanism of neovascularisation in atherosclerotic plaques. J Clin Invest 95: 669-676, 1995.
10. Torzewski, M, Rist, C, Mortensen, RF, Zwaka, TO, Vienek, M, Waltenberger, J, Koenig, W, Schmitz, G, Hombach, V, Tirzewski, J: C-reactive protein in the arterial intima. Role of C-reactive protein receptor-dependent monocyta recruitment in atherogenesis. Arterioscler Thromb Vasc Biol 20: 2094-2099, 2000.
11. Milstien, S, Katusic, Z: Oxidation of tetrahydrobiopterin by peroxynitrite. Biochem Biophys Res Commun 263: 681-694, 1999.
12. Keidar, S, Kaplan, M, Aviram, M: Angiotensin-II-modified LDL is taken up by macrophages via the scavenger receptor, leading to cellular cholesterol accumulation. Arterioscler Thromb Vasc Biol 16: 97-105, 1996.
13. Napoli, C, D'Armiento, FP, Mancini, FP, Postiglione, A, Witztum, JL, Palumbo, G, Palinski, W: Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia: intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest 100: 2680-2690, 1997.
14. Schmidt, AM, Stern, D: Atherosclerosis and diabetes. Curr Atheroscler Rep 2: 430-436, 2000.
15. Steinberg, D, Parthasarathy, S, Carew, TE, Khoo, JG, Witztum, JL: Beyond cholesterol: Modification of low density lipoprotein that increase its atherogenicity. N Engl J Med 320: 915-924, 1989.
16. Betteridge, J: What is oxidative stress? Metabolism 49 (Suppl.2.): 3-8, 2000.
17. Navab, M, Imes, SS, Hama, SY, Hough, GP, Ross, LA, Bork, RW, Valente, AJ, Berliner, JA, Drinkwater, DC, Laks, H, Fogelman, AM: Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J Clin Invest 88: 2039-2046, 1991.
18. Beckman, JA, Creager, MA, Libby, P: Diabetes and atherosclerosis. Epidemiology, pathophysiology and management. JAMA 287: 2570-2581, 2002.
19. Rösen, P, Nawroth, PP, King, G, Moller, W, Tritschler, HJ, Packer, L: The role of oxidative stress in the onset and progression of diabetes and its complications. Diab Metab Res Rev 17: 189-212, 2001.
20. Steinberg, D: Low density lipoprotein oxidation and its pathobiological significance. J Biol Chem 272: 20963-20966, 1997.
21. Hajjar, DF, Haberland, ME: Lipoprotein trafficking in vascular cells: molecular Trojan horses and cellular saboteurs. J Biol Chem 272: 22975-2278, 1997.
22. Libby, P: Molecular bases of the acute coronary syndromes. Circulation 91: 2844-2850, 1995.
23. Paolisso G, Tagliamonte MR, Marfella R, Verrazzo G, D'Onofrio F, Giugliano, D: L-arginine but not D-arginine stimulates insulin-mediated glucose uptake. Metabolism 46: 1068-73, 1997.
24. Ookawara, T, Kawamura, N, Kitagawa, Y, Taniguchi, N: Site-specific and random fragmentation of Cu, Zn superoxide dismutase by glycation reaction. J Biol Chem 267: 18505-18510, 1992
25. Lyons, TJ, Jenkins, AJ: Glycation, oxidation and lipoxidation in the development of the complications of diabetes: A carboxyl stress hypothesis. Diabetes Rev 5: 365-391, 1997.
26. Iso, Y, Kilo, C, Williamson, JR: Cytosolic NADH/NAD+, free radicals and vascular dysfunction in early diabetes mellitus. Diabetologia 40(Suppl.2.): S115-S117, 1997
27. Mangin, EL jr., Kugiyama, K, Nguy, JH, Kerns, SA, Henry, PD: Effects of lysolipids and oxidatively modified low density lipoprotein on endothelium-dependent relaxation of rabbit aorta. Circ Rec 72: 161-166, 1993.
28. Hennes, MM, O'Shaughnessy, IM, Kelly, TM, Labella, P, Egan, BM, Kissebah, AH: Insulin-resistant lipolysis in abdominally obese hypertensive individuals. Hypertension 28: 120-126, 1996.
29. Inoguchi, T, Li, P, Umeda, F, Kakimoto, M, Imamura, M, Aoki, T, Etoh, T, Hashimoto, T, Naruse, M, Sano, H, Utsumi, H, Nawata, H: High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49: 1939-1945, 2000.
30. Libby, P: Current concepts of the pathogenesis of the acute coronary syndromes. Circulation, 104: 365-373, 2001.
31. Hussain, MJ, Peakman, M, Gallati, H, Lo, SS, Hawa, M, Viberti, GC, Watkins, PJ, Leslie, RD, Vergani, D: Elevated serum levels of macrophage-derived cytokines precede and accompany the onset of IDDM. Diabetologia 39: 60-69, 1996.
32. Uemura, S, Matsushita, H, Li, W, Asagami, T, Lee, KH, Harrison, DG, Tsao, PS: Diabetes mellitus enhances vascular matrix metalloproteinase activity. Circ Res 88: 1291-1298, 2001.
33. Kario, K, Matsuo, T, Kobayashi, H, Matsuo, M, Sakata, T, Miyata, T: Activation of tissue factor-induced coagulation and endothelial cell dysfunction in non-insulin-dependent diabetic patients with microalbuminuria. Arterioscler Thromb Vasc Biol 15: 1114-1120, 1995.
34. Fukumoto, H, Naito, Z, Asano, G, Aramaki, T: Immunohistochemical and morphometric evaluation of coronary atherosclerotic plaques associated with myocardial infarction and diabetes mellitus. J Atheroscler Thromb 5: 29-35, 1998.
35. Vinik, AI, Erbas, T, Park, TS, Nolan, R, Pittenger, GL: Platelet dysfunction in type 2 diabetes. Diabetes Care 24: 1476-1485, 2001.
36. Assert, R, Scherk, G, Bumbure, A, Pirags, V, Schatz, H, Pfeiffer, AF: Regulation of protein kinase C by short term hyperglycaemia in human platelets in vivo and in vitro. Diabetologia 44: 188-95, 2001.
37. Li, Y, Woo, V, Bose, R: Platelet hyperactivity and abnormal Ca(2+) homeostasis in diabetes mellitus. Am J Physiol 280: H1480-H1489, 2001.
38. Carr, ME: Diabetes mellitus: a hypercoagulable state. J Diabetes Complications 15: 44-54, 2001.
39. Pickup, JC, Mattock, MB, Chusney, GD, Burt, D: NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 40: 1286-1292, 1997.
40. Warnholtz, A, Nickenig, G, Schultz, E, Macharzina, R, Brasen, JH, Skatchkov, M, Heitzer, T, Stasch, JP, Griendling, KK, Harrison, DG, Bohm, M, Meinertz, T, Munzel, T: Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for the involvement of the renin-angiotensin system. Circulation 99: 2027-2033, 1999.
41. Daemen, MJAP, Lombardi, DM, Bosman, FT, Schwartz, SM: Angiotensin II induces smooth muscle cell proliferation in the normal and injured rat arterial wall. Circ Res 68: 450-456, 1991.
42. Kubo, A, Fukuda, N, Soma, M, Izumi, Y, Kanmatsuse, K: Inhibitory effect of an angiotensin-II type-1 receptor antagonist on growth of vascular smooth muscle cells from spontaneously hypertensive rats. J Cardiovasc Pharmacol 27: 58-63, 1996.
43. Yanagitani, Y, Rakigi, A, Okamura, A, Moriguchi, K, Takiuchi, SM, Oohishi, M, Suzuki, M, Suzuki, K, Higaki, J, Ogihara, T: Angiotensin-II type-1 receptor-mediated peroxide production in human macrophages. Hypertension 33: II335-II339, 1999.
44. Chobanian, AV, Dzau, VJ: Renin angiotensin system and atherosclerotic vascular disease. (In: Fuster, V, Ross, R, Topol, EJ [eds]: Atherosclerosis and coronary artery disease. Vol. I. Philadelphia, Lippincott-Raven, 1996.) pp. 237-242.
45. Naftilan, AJ, Gilliland, GK, Eldridge, CK, Kraft, AS: Induction of the proto-oncogene c-junction by angiotensin II. Mol Cell Biol 10: 5536-5540, 1990.
46. Kim, JA, Berliner, JA, Nadler, JL: Angiotensin-II increases monocyte binding to endothelial cells. Biochem Biophys Res Commun 226: 862-868, 1996.
47. Scharpe, N: The effects of ACE inhibition on progression of atherosclerosis. J Cardiovasc Pharmacol 22(Suppl. 9.): S9-S12, 1993.
48. Ferrario, CM, Deitch, JS, Dean, RH, Strawn, WB: Hypertension and atherosclerosis: a mechanistic understanding of disease progression. Cardiovascular Risk Factors 6: 299-310, 1996.
49. Foris G, Dezső B, Megyesi GA, Füst Gy: Effect of angiotensin II on macrophage functions. Immunology 48: 529-535, 1983.
50. Chobanian, AV: Corcoran lecture: adaptive and maladaptive responses of the arterial wall to hypertension. Hypertension 15: 666-674, 1990.
51. Potter, DD, Sobey, CG, Thompkins, PK, Rossen, JD, Heistad, DD: Evidence that macrophages in atherosclerotic lesions contain angiotensin II. Circulation 98: 800-807, 1998.
52. Strawn, WB, Chappell, MC, Dean, RH, Kivlighn, S, Ferrario, CM: Inhibition of early atherogenesis by losartan in monkeys with diet-induced hypercholesterolemia. Circulation 101: 1586-1593, 2000.
53. Herren, B, Raines, EW, Ross, R: Expression of a disintegrin-like protein in cultured human vascular cell and in vivo. FASEB J 11: 173-180, 1997.
54. Mancini, GBJ, Henry, GC, Macaya, C, O'Neill, BJ, Pucillo, AL, Carere, RG, Wargovich, TJ, Mudra, H, Lüscher, TF, Klibaner, MI, Haber, HE, Uprichard, ACG, Pepine, CJ, Pitt, B: Angiotensin-converting enzyme inhibition with quinapril improves endothelial vasomotor dysfunction in patients with coronary artery disease. The TREND (Trial on Reversing Endothelial Dysfunction) study. Circulation 94: 258-265, 1996.
55. Powell, JS, Clozel, JP, Muller, RKM, Kuhn, H, Hefite, F, Hosang, M, Baumgartner, HR: Inhibitors of angiotensionconverting enzyme prevent myointimal proliferation after vascular injury. Science 245: 186-188, 1989.
56. Ridker, PM, Hennekens, CH, Buring, JE, Rifai, N: Creactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 342: 836-843, 2000.
57. Xing, Z, Gauldie, J, Cox, G: IL-6 is an antiinflammatory cytokine required for controlling local and systemic acute inflammatory responses. J Clin Invest 101: 311-320, 1998.
58. Lemieux, I, Pascot, A, Prud'homme, D, Alméras, N, Bogaty, P, Nadeau, A, Bergeron, J, Després, JP: Elevated C-reactive protein. Another component of the atherothrombotic profile of abdominal obesity. Arterioscl Thromb Vasc Biol 21: 961-967, 2001.
59. Ridker, PM: High-sensitivity C-reactive protein. Potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 103: 1813-1818, 2001.
60. Ridker, PM, Cushman, M, Stampfer, M, Tracy, R, Hennekens, CH: Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 336: 973-979, 1997.
61. Jialal, I, Stein, D, Balis, D, Grundy, SM, Adams-Huet, B, Devaraj, S: Effect of hydroxymethyl glutaryl coenzyme A reductase inhibitor therapy on high sensitive C-reactive protein levels. Circulation 103: 1933-1935, 2001.
62. Horne, BD, Muhlestein, JB, Carlquist, JF, Bair, TL, Madsen, TE, Hart, NL, Anderson, JL: Statin therapy, lipid levels, C-reactive protein and the survival of patients with angiographically severe coronary artery disease. J Am Coll Cardiol 1774-1780, 2000.
63. Ridker, PM, Rifai, N, Clearfield, M, Downs, JR, Weis, S, Miles, JS, Gotto, AM, Phil, D: Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 344: 1959-1966, 2001.
64. Ichiki, T, Takeda, K, Zokunou, T, Iino, N, Egashira, K, Shimokawa, H, Hirano, K, Kanaide, H, Takeshita, A: Downregulation of angiotensin II type 1 receptor by hydrophobic 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 21: 1896-1901, 2001.
65. Heilbronn, LK, Noakes, M, Clifton, PM: >Energy restriction and weight loss on very-low-fat diets reduce C-reactive protein concentrations in obese, healthy women. Arterioscler Thromb Vasc Biol 21: 968-970, 2001.
66. Hsueh, W, Law, RE: PPAR-gamma and atherosclerosis. Effect on cell growth and movement. Arterioscler Thromb Vasc Biol 21: 1891-1895, 2001.
67. Marx, N, Sukhova, GK, Collins, T, Libby, P, Plutzky, J: PPAR-alpha activarors inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation 99: 3125-3131, 1999.
68. Beckman, JA, Goldfine, AH, Gordon, MB, Garrett, LA, Creager, MA: Inhibition of protein kinase C beta prevents impaired endothelium-dependent vasodilation caused by hyperglycemia in humans. Circ Res 90: 107-111, 2002.
- Közlésre érkezett: 2002. augusztus 13..
- Közlésre elfogadva: 2002. október 19.
- A szerző levelezési címe: Dr. Pogátsa Gábor
- 1539 Budapest, Postafiók 561