Semmelweis Orvostudományi Egyetem, I. Belgyógyászati Klinika
LIPIDANYAGCSERE-ZAVAROK CUKORBETEGSÉGBEN I. A DIABETESRE JELLEMZÖ LIPIDELTÉRÉSEK ÉS AZOK PATOMECHANIZMUSA*
Gerő László dr.
Összefoglalás
Szerző a cukorbetegségben leggyakrabban előforduló lipidanyagcsere-zavarokat tekinti át. Tárgyalja az inzulin-dependens és a nem-inzulin-dependens típusban észlelt lipideltéréseket, azok kialakulásának pathomechanizmusát, és ezen eltérések kapcsolatát a haemostasis, ill. az endothelfunkció zavaraival. Végül röviden összefoglalja ezen eltérések komplex szerepét a thrombo-atherogen folyamatokban.
Kulcsszavak: diabetes mellitus, metabolikus szindróma, lipidanyagcsere-zavarok, atherosclerosis
Disorders of lipid metabolism in diabetes mellitus. 1. The characteristic lipid alterations and their pathomechanism
Summary
The author gives an account of the disturbances in lipid metabolism most frequently seen in diabetes. The pathomechanism of these lipid disorders in type 1 and type 2 diabetes, und the connections between lipid abnormalities and altered haemostatic and endothelial functions are also discussed. Finally, a short summary is given regarding the complex role of lipid alterations in the vascular thromboatherogen process.
Key words: diabetes mellitus, metabolic syndrome, disorders in lipid metabolism, atherosclerosis
*Az összefoglaló munka terápiás lehetőségeket taglaló második részét következő számunkban jelentetjük meg.
A dolgozat azokat a lipidanyagcsere-zavarokat foglalja össze, amelyek a leggyakrabban észlelhetök cukorbetegeken. Tisztában kell lennünk természetesen azzal a ténnyel, hogy az alább ismertetendő lipideltérések más klinikai állapotokban is előfordulhatnak, azaz nem teljesen specifikusak. Mégis, a cukorbetegségben való gyakori megjelenésük miatt megengedhető a "diabetesre jellemző" megjelölés.
I. A lipidanyagcsere zavarai inzulin-dependens diabetesben (IDDM)
Jól beállított IDDM-es betegekben gyakorlatilag nem több a lipideltérés mint a nem-diabeteses populációban.
Érdekes módon a HDL-cholesterin-szint IDDM-ben még a normálisnál magasabb is lehet, ennek biológiai jelentősége azonban kérdéses, hiszen korai atheroscleroticus elváltozások ennek ellenére kifejlődnek.
A szénhidrát-anyagcsere akut kisiklásakor, diabeteses ketoacidosisban a VLDL és a chylomikron szintjének jelentős emelkedése alakulhat ki, amely azonban az anyagcsere rendezésével párhuzamosan megszűnik. Tartósan rossz beállítás esetén mind a cholesterin, mind a triglycerid koncentrációja emelkedhet a szérumban. A szénhidrát-anyagcsere rendezésével mindezen lipideltérések normalizálódnak.
Egyes megfigyelések szerint a lipoprotein(a)/Lp(a)/ szintje emelkedett volt IDDM-es betegeken, mások ezt nem erősítették meg.1 Jenkins és mtsai, valamint Groop és mtsai szerint az Lp(a) szintje IDDM-ben csak macroalbuminuria esetén volt emelkedett – kérdéses tehát, hogy az észlelt emelkedés a cukorbetegség vagy a nephropathia (fehérjevesztés) következménye volt-e.2,3
II. A lipidanyagcsere zavarai nem-inzulin-dependens diabetesben (NIDDM)
NIDDM-ben gyakori és típusos lipideltérések figyelhetök meg. Mivel ezek az eltérések általában már a diabetes manifesztációja előtt is kimutathatók, helyesebb ezeket úgy tekinteni, mint a metabolikus szindróma jellegzetes lipideltéréseit. Ilyen eltérések (1) az elhúzódó postprandialis lipaemia (nagyrészt a lipoproteinlipáz ((LpL)) enzim elégtelen aktivitása miatt), (2) a fokozott hepatikus VLDL-szekréció, (3) az LDL kvalitatív eltérései, (4) a magas triglycerid- és az (5) alacsony HDL-szint.
(1) Az LpL elégtelensége – következményes postprandialis hyperlipidaemia
Táplálékfelvételt követően a bélben lebontott és a bélfalban reszintetizálódó triglyceridek (TG) és cholesterin-észterek a bélsejtekben termelődő Apo-B48 (kisebb részben Apo-AI és -AIV) apoproteinnel egyesülve ún. naszcens chylomikront képeznek és a ductus thoracicuson át jutnak a keringésbe, ahol később még Apo-CII és -CIII, ill. Apo-E fehérjékkel egészülnek ki. Eliminációjuk az LpL aktivitásához kötött. Az enzim a kapillárisok falában, az endothelsejteken található, s egyik fő feladata (a VLDL hidrolízise mellett), hogy étkezést követően a chylomikronokból triglyceridet hasítson le. Így a chylomikron részecskék egyre kisebbé válnak, TG-tartalmuk csökken, ugyanakkor a sejtek (elsősorban a zsír- és izomsejtek) számára szabad zsírsav és glycerin keletkezik. Eközben a részecskék felszíne is fokozatosan módosul, az említett apoproteinek közül egyre inkább az Apo-E válik a felszínen "szabaddá". Amikor a chylomikron részecskék TG-tartalma az eredetinek már csak kb. 20%-a, további hasításuk leáll, és a maradék – ún. chylomikron-remnant – részecskék a májba kerülnek. Ott a májsejtek felszínén lévő Apo-E-specifikus remnant-receptorokhoz (LRP) kötödnek, majd endocitózissal a májsejtekbe jutva a lizoszómákban lebontódnak.
Az LpL-t aktuálisan az Apo-CII aktiválja, de az enzim aktivitását az inzulin is szabályozza. Inzulinhiány, ill. a hormon hatástalansága esetén a vér TG-szintje jelentősen emelkedhet. Ilyen eltérés akutan is kialakulhat diabeteses ketoacidosisban, ill. kómában. Fontosabb ennél, hogy inzulinrezisztens állapotban az étkezést követően jelentős és elhúzódó lipidaemia – döntöen hypertriglyceridaemia fejlődhet ki.
Az elhúzódó postprandialis triglyceridaemia kialakulásának patomechanizmusa meglehetösen bonyolult, és részleteiben mind a mai napig nem teljesen tisztázott. Bármennyire kézenfekvönek látszana, hogy az étkezést követő magas TG-koncentráció teljes egészében a táplálékból felszívódó TG-böl, annak elégtelen bontásából származik, a vizsgálatok nem ezt igazolták: Gernest és mtsai,4 valamint Schneeman és mtsai azt találták, hogy lipidterhelést követően a szérumban a TG-dús lipoproteinekben az Apo-B48 mellett jelentős mennyiségben Apo-B100 protein volt kimutatható, vagyis a TG-dús lipoproteinek nem csupán intestinalis, hanem jelentős részben májeredetű (VLDL) apoproteint és triglyceridet is tartalmaztak. Ennek az a magyarázata, hogy – szemben a normális anyagcseréjű egyénekkel, akiken az LpL kapacitása messzemenöen elegendő a postprandialis időszakban is a chylomikron-TG és a VLDL-TG hidrolíziséhez – az inzulinrezisztens állapotokban az LpL aktivitása csökkent, és így zsírfogyasztát követően a chylomikronok telítik az enzimet. Mivel az LpL preferenciálisan bontja a chylomikronokat, ezért a VLDL hidrolízise átmenetileg lelassul. Így a postprandialis TG-szint-emelkedés a chylomikron-rem-nant-TG mellett jelentős részben a VLDL-TG-böl is származik. Ezt a hipotézist tovább erősíti az a megfigyelés, hogy zsírterhelést követően az IDL Apo-B100 tartalma szignifikánsan csökkent a lipaemia idején, azaz a VLDL metabolizmusa (a VLDL?IDL?LDL átalakulás) lelassult ezen periódus alatt.6 Tovább növekszik a VLDL vérszintje amiatt is, hogy az inzulin nem képes megfelelően szupprimálni a máj VLDL-termelését (lásd lejjebb). Megnő az LDL és a HDL frakciók triglycerid-tartalma is. Ugyanakkor, a keringésben való hosszabb tartózkodás során – a cholesterin-ester-transzferprotein (CETP) aktivitása folytán – az eredetileg TG-dús részecskék cholesterin-észterben is egyre gazdagabbá válnak: a HDL-2-röl a chylomikron-remnant és a VLDL-IDL részecskékre cholesterin-észter kerül, TG-tartalmuk egy részét viszont a HDL-2 veszi át. Utóbbit a hepatikus lipáz HDL-3-má alakítja, így a szérum HDL-2 tartalma csökken. A postprandialis hyperlipidaemia tehát egy komplex anyagcsere-kisiklás, amely gyakorlatilag az összes lipoprotein frakciót érinti, azok mennyisége és összetétele atherogen irányba alakul át.7,8,9,10
Metabolikus szindrómában szenvedő egyéneken standard lipidterhelés során két jól elkülönülő alcsoportot találtak: az egyik alcsoportban a TG-emelkedés mérsékelten volt magasabb a normális kontrollcsoporténál, míg a betegek egy kisebb hányadán ennél szignifikánsan magasabb emelkedést észleltek (ún. "TG high-responders"). Érdekes módon, ilyenfajta különbség a metabolikus szindrómában szenvedő szülök egészséges (normális testsúlyú, oralis glukózterhelés (oGT) alapján nem-diabeteses, normális éhomi lipidszintekkel rendelkezö) leszármazottain, söt, a fenti familiáris terheltségtöl mentes egészséges egyedek egy kis csoportján is kimutatható volt.11 Ugyanakkor a "TG high-responder" egyéneken szignifikánsan magasabb inzulinszinteket találtak.11,12
Mindez amellett szól, hogy a kóros postprandialis TG-emelkedés az inzulinrezisztencia egyik legkoraibb metabolikus megnyilvánulása lehet – korábbi, mint a glukózintolerancia.
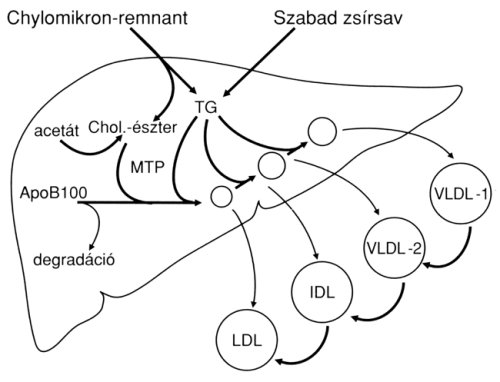
1. ábra: Az Apo-B100 lipoproteinek hepatikus szintézise. A máj két forrásból is kap szubsztrátot a lipoprotein-szintézishez: a táplálékból keletkező chylomikron-remnant és a perifériás zsírszövet lipolízise során keletkező szabad zsírsav egyaránt fokozza a lipoprotein-szintézist (hiányukban az Apo-B100 molekula intracellulárisan degradálódik). A kétféle lipidvegyület az MTP (mikroszomális transzfer protein) közvetítésével egyesül a Apo-B100 fehérjével. A beépített lipidek mennyiségétöl és minőségétöl függ a keletkezett lipoprotein nagysága és denzitása. Ugyanakkor, a nagy mennyiségben keletkező VLDL-1 TG-tartalmának fokozatos hidrolízise az ismert VLDL-1 VLDL-2 IDL LDL átalakuláshoz vezet (ún. Apo-B100 kaszkád).
Az elhúzódó postprandialis hyperlipidaemiának egyre nagyobb szerepet tulajdonítunk a korai atherosclerosis kifejlődésében. A postprandialis TG-szintek szorosabban korrelálnak a coronariabetegség gyakoriságával, mint az éhomi szintek.7 Philips és mtsai követéses vizsgálata szerint a coronariasclerosis progressziója elsősorban a remnant-TG-szintekkel mutatott korrelációt.12 Más vizsgálatokban szoros összefüggés mutatkozott az alimentaris lipaemia mértéke és az angiographiával kimutatott coronariasclerosis súlyossága között. Mindez egyértelműen igazolja a postprandialis hyperlipidaemia elsörendű atherogen szerepét.
Az idézett adatok tükrében fontos hangsúlyozni azt a tényt, hogy a postprandialis hyperlipidaemia nem rövid ideig fennálló állapot. Amíg a postprandialis hyperglykaemia másfél–két óra alatt lezajlik, az étkezés vagy lipidterhelés utáni TG-szint-emelkedés akár 12 órán túl is elhúzódhat, azaz inzulinrezisztens egyéneken a kóros TG-emelkedés, s a következményes atherogen lipidprofil a nap nagyobb felében fennállhat. Érthető tehát, hogy a coronaria-betegség, ill. az egyéb vascularis szövődmények a postprandialis TG-szintekkel szorosabb korrelációt mutatnak mint az éhomi TG-szinttel. A veszélyeztetett egyedek kiszűrésére egyre elterjedtebben alkalmaznak különböző lipidterheléseket (ezek lipidősszetételére, kalóriatartalmára, szénhidrátmennyiségére vonatkozóan sajnálatos módon nincs elfogadott nemzetközi standard). A terheléssel kiszürhetök a csökkent TG-toleranciát mutató személyek (inzulinrezisztenciában a csökkent TG-tolerancia sokkal korábban kialakulhat, mint a csökkent glukóztolerancia). A fenti adatok alapján nemcsak a "high responder" egyének, de már az ún. csökkent TG-toleranciát mutató személyek korai diétás, szükség esetén gyógyszeres kezelése is igen fontos lenne.
(2) Fokozott hepatikus VLDL-szekréció
A VLDL komplexum Apo-B100 protein és triglycerid (kisebb mennyiségben cholesterin és cholesterin-észter, valamint foszfolipid) komponensekböl áll. Az Apo-B100 protein a májsejtekben képződik, szintézisének elindításához kis mennyiségű TG vagy szabad zsírsav, esetleg cholesterin jelenléte szükséges. A hepatocelluláris VLDL-produkció két fő forrása a remnant-triglycerid és a zsírszövetek lipolíziséböl származó szabad zsírsav. Diabetesben mindkét szubsztrát nagyobb mennyiségben áll rendelkezésre (utóbbi vegyület azért, mert az inzulin nem képes a szöveti lipolízist szupprimálni), és a fokozott szubsztrát-kínálat a májbeli VLDL-szintézis növekedéséhez vezet (1. ábra). Szignifikáns korreláció mutatható ki a szérum inzulinszintje és a VLDL-képzés mértéke között, ezért a metabolikus szindrómában észlelt fokozott VLDL-produkciót sokan a hyperinsulinaemia direkt hatásának tulajdonítják.
A VLDL komplexum egy nagyobb VLDL-1 (60–400 Sf) és egy kisebb VLDL-2 (20–60 Sf) összetevöre választható szét. Taskinen és mtsai NIDDM-es betegeken inzulin infúzió adása során mérték a hepatikus VLDL-szekréciót, és azt találták, hogy amíg a kontrollcsoportban az inzulin szignifikánsan (mintegy 50%-kal) csökkentette a VLDL-1-szintézist, addig a cukorbeteg-csoportban ez a csökkenés nem következett be. A VLDL-2 szintézisében nem mutatkozott ilyen különbség.13 Az eredményekböl egyrészt az következik, hogy a VLDL-1 és -2 képzésének regulációja eltérő, másrészt azonban az a fontos tény is, hogy a metabolikus szindrómában észlelt fokozott VLDL-termelés nem a magas inzulinszintnek, hanem éppen az inzulinhatás elmaradásának, az inzulinrezisztenciának a következménye (ugyanúgy, mint pl. a fokozott hepatikus glukózprodukció). A TG-ben dúsabb VLDL-1 viszont atherogen hatású, felszaporodása az atherosclerosis fokozódásához vezet.
(3) Az LDL kvalitatív változásai
Már a hetvenes évek közepén ismert volt, hogy a szérumban található LDL-partikulumok – az alkalmazott szeparációs módszertöl függöen – három vagy annál több frakcióra választhatók szét. Ezen összetevök nagysága, kémiai összetétele, ill. denzitása finoman különbözik egymástól. Az LDL-1 frakció denzitása a legkisebb, az LDL-3-é a legnagyobb. A részecskék nagysága fordítottan arányos a denzitással, ennek alapján az LDL-3-t "small dense LDI:"-nek nevezi az irodalom.14,15 Érdekes, hogy az egyes frakciók kialakulásának aránya a szérum triglyceridtartalmától függ: növekvő TG-szint mellett egyre nő a kisebb méretű, de nagyobb denzitású partikulumok aránya16 (1. táblázat). A különböző LDL-részecskék előfordulási aránya alapján a vizsgált személyeket két csoportba lehetett sorolni: az egyikben – normális TG-szint mellett – az LDL-1 dominált (ún. "A típus"), a másikban – emelkedett TG-szint mellett – az LDL-3 (ún. "B típus", 2. ábra).
| LDL frakció | Denzitás (g/ml) |
Részecske- átmérő (nm) |
Szérum- TG-szint (mmol/l) |
| LDL-1 | 1,020-1,035 | 260-270 | 1,0-1,3 |
| LDL-2 | 1,035-1,045 | 250-260 | 1,3-1,5 |
| LDL-3 | 1,045-1,060 | < 250 | > 1,5 |
1. táblázat: Összefüggés az LDL-partikulumok nagysága, denzitása és a szérum TG-szintje között
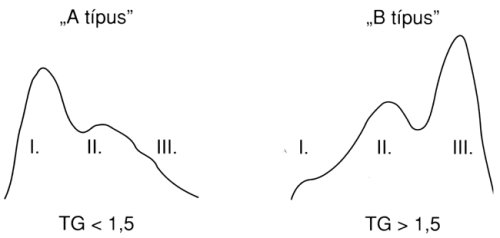
2. ábra: Az LDL komplexum (legalább) három komponensre bontható. Alacsony TG-szint mellett az LD-l ("A"-típus), míg magas TG-szint mellett az LDL-3 frakció dominál (ún. "B"-típus). Utóbbi frakció kevésbé kötödik a hepatikus LDL-receptorokhoz, ugyanakkon könnyebben oxidálódik. Felszaporodása végül is a habos sejtek (foam cells) mennyiségének növekedéséhez, és az atheroscleroticus folyamat felgyorsulásához vezet.
Az össz-LDL-cholesterin szintje metabolikus szindrómában nem változik szignifikánsan. Ugyanakkor az LDL-partikulumok nagyságbeli elemzése alapján több szerző egybehangzóan igazolta, hogy mind manifeszt NIDDM-ben, mind metabolikus szindrómában a small dense LDL részecske fordul elő nagyobb arányban, azaz a betegek többsége a B típusba sorolható.17,18,19
A small dense LDL eliminációja a plazmából két és félszer lassúbb, mint az LDL-I-é, oxidációja viszont lényegesen gyorsabb.20 Ezenkívül az LDL oxidációja a vércukorszint növekedésével párhuzamosan fokozódik. Normális lipidanyagcsere esetén az LDL döntő többsége (kb. 70%-a) a máj LDL-receptoraihoz kötödik és az epébe, ill. epesavként a bélbe választódik ki. A small dense LDL, ill. annak oxidált formája viszont nem, vagy alig képes kötödni a májbeli LDL-receptorokhoz, ezért a monocyta-macrophag rendszer ún. scavenger (eltakarító) receptoraihoz kötödik, és azokkal együtt az érfalban halmozódik fel.
További lényeges különbség a LDL-3 katabolizmusában, hogy amíg az Apo-B- és -B/E-receptorokon át internalizálódó LDL-I-cholesterin mennyiségével arányosan csökkenti a sejtmembránon lévő LDL-receptorok számát, valamint gátolja a sejtekben a HMG-CoA reduktáz enzimet, és így a sejt két úton is védekezni képes a túlzott intracelluláris cholesterin-felhalmozódással szemben, addig a scavenger receptorokon át a sejtekbe kerülő cholesterin nem fejt ki ilyenfajta gátló hatást, emiatt a monocyta-macrophag sejtek túltelítödnek cholesterinnel és létrejönnek a habos sejtek (foam cell). E sejteknek az érfalban történő felhalmozódásával kialakul az ún. fatty streak ("zsírcsík"), amely a további thrombo-atherogen folyamatok kiindulási pontja. Nem meglepő tehát, hogy azokban a betegcsoportokban, amelyekben a small dense LDL dominanciáját észlelték (B típus) a coronariabetegség rizikójának 3–7-szeres növekedését mutatták ki, s ez független volt az össz-LDL-cholesterin szintjétöl.21,22
A metabolikus szindromában észlelt magas coronaria-halálozás tehát részben az LDL atherogen típusának felszaporodásával, a "B" típus dominanciájával függhet össze.
(4) Emelkedett TG-szint
Metabolikus szindrómában, ill. NIDDM-es betegekben a legáltalánosabb, leggyakrabban regisztrált lipideltérés a TG-szint emelkedése. A TG – a glukóz mellett – a sejtek fő energiaforrása. A vérben elsősorban a chylomikron, a VLDL- és az IDL-részecske alkotórészeként kering, de kisebb mennyiségben, kb. 10–15%-ban az LDL frakcióban és mintegy 5–10%-ban a HDL-2 és HDL-3 lipoproteinekben is megtalálható. Metabolikus szindrómában a TG-szint emelkedésének oka elsősorban a TG-ben dús lipoproteinek (chylomikron, VLDL, IDL) vérkoncentrációjának emelkedése, de a többi lipid frakció összetétele is megváltozik, TG-tartalmuk nő.
A TG-szint és a metabolikus szindróma számos egyéb jellemző paramétere között szoros kapcsolat mutatható ki: nagy betegcsoportokon vizsgálva a szérum TG-szintje statisztikailag szignifikánsan korrelált a clamp-technikával mért inzulinrezisztencia mértékével,23 a BMI-vel, a derék/csípő aránnyal,24 a small dense LDL-részecskék előfordulási gyakoriságával,25 a szérum inzulin-szinttel, a plazminogénaktivátor-inhibítor-1 (PAI-1) koncentrációjával és fordítottan korrelált a HDL-szinttel. A szérum TG-szintje fontos prediktív mutatója a cardiovascularis morbiditásnak és mor-talitásnak.26,27 (Ez nem mond ellent annak a ténynek, hogy a postprandialis TG-szint még erősebben korrelál a vascularis megbetegedésekkel, mint az éhomi szint).
A magas TG-szint atherogen hatásának patomechanizmusa a fent leírtakból (VLDL-1 és small dense LDL felszaporodása, fokozott lipidoxidáció, preferenciális kötödés a scavenger receptorokhoz, fatty streak kialakulása stb.) jól érthető. Hangsúlyozni kell azonban, hogy a magas TG-szinttel járó állapotokban még egy másfajta káros vascularis hatás is érvényesül, és ez a fokozott thrombosis-hajlam. Az emelkedett PAI-1-szintröl már volt szó. A magas TG-szinttel párhuzamosan emelkedik a VII. alvadási faktor koncentrációja, és fokozódik a VII?VII-foszfolipid-komplex átalakulás, azaz a VII. faktor aktív formájának keletkezése.28,29 Növekszik a fibrinogén- és a thrombingeneráció, ami részben a fokozott lipidperoxidáció következménye.30,31 A magas TG-szint tehát egyúttal ún. prokoaguláns állapotot is eredményez, ami tovább növeli a cardiovascularis betegségek rizikóját.
(5) Alacsony HDL-szint
A HDL a legkisebb, legnagyobb sürüségű lipoprotein, feladata az ún. fordított cholesterintranszport, azaz a cholesterin felvétele és visszaszállítása a perifériás szövetekböl (részben az érfalból is) a májba. Ez a feladat alapvető az egész szervezet cholesterin-egyensúlya szempontjából, mivel szervezetünk egyetlen sejtje sem képes a cholesteringyürű bontására. A máj viszont részben az epébe, részben epesav formájában a bélbe kiválasztja a visszaszállított cholesterint, ezáltal biztosítva a "felesleg" eltávolítását. Ezenkívül a HDL gátolni képes az LDL érfalba történö lerakódását, ill. oxidációját.32,33 Számos experimentális és klinikai epidemiológiai adat szól amellett, hogy a HDL gátolja az atherosclerosis folyamatát.
A HDL különböző szubtípusainak keletkezése és ezen szubtípusok egymásba történő átalakulása [(1) pont] meglehetösen bonyolult és komplex folyamat.34,35,36 Tény, hogy a metabolikus szindrómára ezen protektív cholesterin-frakció (ezenbelül elsősorban a fő védöfrakciónak tartott HDL-2, ill. a kizárólag Apo-AI-et tartalmazó partikulumok) kifejezett csökkenése a jellemző. A HDL-szint negatívan korrelál a TG-szinttel, a centrális obesitas mértékével és az inzulinszinttel de pozitívan korrelál az inzulinszenzitivitással.37 Tekintettel a TG-szint és a HDL-szint közötti szignifikáns negatív korrelációra, ellentmondó adatokat közöltek arra vonatkozóan, hogy metabolikus szindrómában az alacsony HDL- vagy a magas TG-szint tekinthetö-e a fő cardiovascularis rizikótényezőnek. Egy multivariáns logaritmikus regresszióanalízis szerint mind a magas TG-, mind pedig az alacsony HDL-2-szint önálló, független rizikótényezőnek tekinthető.7
* * *
Az ismertetett eltérések alapján nyilvánvaló, hogy metabolikus szindrómában a lipidanyagcsere atherogen irányba tolódik el. A magas (éhomi és/vagy postprandialis) TG-szint, a fokozott VLDL- (ezen belül elsősorban a VLDL-1-) szekréció, a small dense LDL nagyobb arányú képződés végül is a lipidoxidáció fokozódásához vezet. A magasabb vércukorszint miatt pedig nő a glikált lipoproteinek aránya, amelyek szintén fokozottan hajlamosak az oxidációra.38 E kóros, ill. módosult lipoproteinek nem a normális metabolikus úton, a máj LDL-receptorain át távolítódnak el a keringésböl, hanem a macrophagok scavenger receptoraihoz kötödnek.39
A macrophagok mellett az érfal módosult simaizomelemei is nagy mennyiségben vesznek fel cholesterint. Utóbbiak az érfal simaizomrétegéböl származnak, és a magas inzulinszint hatására egyrészt proliferálódnak, másrészt bevándorolnak a tunica mediából az intimába, ily módon jönnek létre az ún. myointimalis sejtek (egyes vizsgálatok szerint az inzulin külön-külön receptorokon fejti ki metabolikus és proliferatív hatását, emiatt a proliferatív hatás inzulinrezisztenciában is érvényesül). Az intimában felhalmozódott, cholesterinnel túltelített sejtekböl alakul ki az atheroma. Az alacsony HDL-szint miatt a cholesterin eltávolítása az érfalból lelassul, a primer plaque egyre nő. Az oxidált LDL ugyanakkor toxikus hatást fejt ki az endothelsejtekre. Normális körülmények között az endothel böven termel olyan anyagokat, amelyek egyrészt értágító hatásúak (nitrogén-monoxid, prostacyclin), másrészt gátolják a thrombocyták és a monocyták érfalhoz történő vándorlását, ill. a simaizom-proliferációt. E faktorok egyensúlyban vannak a vasoconstrictiót és thrombocyta-aggregációt fokozó tényezőkkel. Az oxidált LDL hatására a vasodilatativ endothelfunkciók károsodnak, és túlsúlyba kerülnek a vasoconstrictor tényezők (endothelin, thromboxan-A2, prostaglandin-H2). Ugyanakkor az oxidált LDL maga is kemotaktikus hatású, és stimulálja a monocyta-macrophag sejtek, ill. a simaizomelemek migrációját. Így egyrészt fokozódik a vasoconstrictio, másrészt az érfalhoz aggregálódott thrombocyták az ér lumene felöl, a növekvő atheroma pedig az érfal felöl szükíti egyre tovább az átmérőt.40 Mivel a haemostasis egyensúlya az alvadás irányába tolódott el, az aggregálódott thrombocyták halmaza egyre nő. A puha primer plaque (ún. soft plaque) felett az egyre nagyobb mechanikai hatásnak kitett szűkület helyén végül is előbb vagy utóbb bereped az érfal, és a fissura helyén thrombosis alakul ki, ami az érfal teljes elzáródásához vezethet. Amint a leírásból nyilvánvaló, a metabolikus szindróma atherogen történései az atherosclerosis több nagy elméletét (lipid-teória, thrombogen-teória, érfal-proliferáció, endothel dysfunctio stb.) is összekapcsolják. Mindez érthetövé teszi a metabolikus szindrómában szenvedökön kimutatott emelkedett cardiovascularis morbiditást és mortalitást, és minél korábbi, erélyes lipidcsökkentő kezelést tesz szükségessé.
IRODALOM
1. Dubrey, SV, Reaweley, DR, Seed, M: Risk factors for cardiovascular disease in IDDM. Diabetes 43: 831–835, 1994.
2. Jenkins, AJ, Steele, JS, Janus, ED, Best, JD: Increased plasma apolipoprotein (a) levels in IDDM patients with microalbuminuria. Diabeles 40: 787–790, 1991.
3. Groop, PH, Viberti, GC, Elliott, TG, Friedman, R, Mackie, A, Ehnholm, C, Jauhiainen, M. Taskinen, MR: Lipoprotein(a) in type 1 diabetic patients with renal disease. Diabetic Medicine 11: 961–967, 1994.
4. Genest, J, Sniderman, A, Cianflone, K, Teng, B, Wacholder, S, Marcel, Y. Kwiterovich. P: Hyperapobetalipoproteinemia: plasma lipoprotein responses to oral fal load. Arteriosclerosis 6: 297–304, 1986.
5. Scheeman, BO, Kotite, L, Todd, KM, havel JR: Relationships between the responses of triglyceride-rich lipoproteins in blood plasma containing apoliporpoteins B48 and B100 to a fat-containing meal in normolipidemic humans. Proc Natl Acad Sci USA 90: 2069–2073, 1993.
6. Karpe, F, Steiner, G, Olivecrona, T, Carlson, LA. Hamsten, A: Metabolism of triglyceride-rich liporpoteins during alimentary lipemia. J Lipid Res 29: 925–936, 1988.
7. Patsch, JR, Miesenböck, G, Hopferwieser, T, Mühberger, V, Knapp, E, Dunn, JK, Gotto, AM Jr, Patsch, W: Relation of triglyceride metabolism and coronary artery disease: studies in the prostprandial state. Arterioscler Thromb 12: 1336–1345, 1992.
8. Zilversmit, DB: Atherogenesis: a postprandial phenomenon. Circulation 60: 473–485, 1979.
9. Kirchmair, R, Ebenbichler, CF, Patsch, JR: Post-prandial lipaemia. (In: Betteridge DJ., Bailliere Tindall, and Dyslipidaemia. Bailliere's Clinical Endocrinology Metabolism, ed. 1995) pp. 705–720.
10. Chen, YD, Swami, S, Skowronski, R. Coulston, A, Reawen, GM: Differences in postprandial lipemia between patients with normal glucose tolerance and non-insulindependent diabetes mellitus. J Clin Endocrin Metab 76: 172–177, 1993.
11. Schrezenmeir, J, Keppler, I, Fenselau, S, Weber, P, Biesalski, HK, Probst, PR, Laue, C, Zuchhold, HD, Prellwitz, W, Beyer, J: The phenomenon of high-triglyceride response to an oral lipid load in healthy subjects and its link to the metabolic syndrome. Ann N Y Acad Sci 683: 302–314, 1993.
12. Philips, NR, Waters, D, Havel JR: Plasma lipoproteins and progression of coronary artery disease evaluated by angiography and clinical events. Circulation 88: 2762–2770, 1993.
13. Malmström, K, Packard, JC, Caslake, M, Bedford, D. Stewart, P, Yki-Jarvinen, H. Shepherd, J, Taskinen, MR: Defective regulation of triglyceride metabolism by insulin in the liver in NIDDM Diabetologia 40: 454–462, 1997.
14. Goebel, R, Garnick, M, Berman, M: A new model for low density apoprotein kinetics: evidence for two labelled moieties. Circulation 54 (Suppl): 11–14, 1976.
15. Krauss, RM, Burke, DJ: Identification of multiple subclasses of plasma low density lipoproteins in normal humans. J Lipid Res 23: 97–104, 1982.
16. McKeone, BJ, Patsch, JR, Pownall, HJ: Plasma triglycerides determine low density lipoprotein composition, physical properties and cell specific binding. J Clin Invest 91: 1926–1933, 1993.
17. Feingold, KR, Grunfeld, C, Pang, M, Dorrler, D, Krauss, KM: LDL subclass phenotypes and triglyceride metabolism in non-insulin dependent diabetes. Arterioscler Thromh 12: 1496–1502. 1992.
18. Reaven, GM, Chen, YD, Jeppesen, J, Maheux, P, Krauss, RM: Insulin resistance and hyperinsulinemia in individuals with small, dense, low density lipoprotein particles. J Clin Invest 92: 141–146, 1993.
19. Detwart, MW, Laker, MF, Dyer, RG, Game, F, Mitcheson, J, Winocour, PH, Alberti, KGMM: Lipoprotein compositional abnormalities and insulin resistancc in Type II diabetic patients with mild hyperlipidemia. Arterioscler Thromb 13: 1l46–1052, 1993.
20. De Graaf, J, Huk-Lemmers, HLM, Hectors, PNM, Demacker, PNM, Hendricks, JCM, Stalenhoef, AFH: Enhanced susceptibility to in vitro oxidation of the dense low density lipoprotein subfractions in healthy subjects. Arterioscler Thromb 11: 298–306, 1991.
21. Austin, MA, Breslow, JL, Hennekens, CH,. Buring, JE, Willet, WC, Krauss, RM: Low density lipoprotein subclass patterns and risk of myocardial infarction. JAMA 260: 1917–1921, 1988.
22. Griffin, BA, Freeman, DJ, Tait, GW: Role of plasma triglyceride in the regulation of plasma low density lipoprotein (LDL) subfractions: relative contribution of small, dense LDL to coronary heart disease risk. Atherosclerosis 106: 241–253, 1994.
23. Reaven, GM: Role of insulin resistance in human disease (syndrome X): an expanded definition. Annu Rev Med 44: 121–131, 1993.
24. Sorge, F, Schwartzkopf, W, Neuhaus, GA: Insulin response to oral glucose in patients with a previous myocardial infarction and in patients with peripheral vascular disease: hyperinsulinism and its relationships to hypertriglyceridemia and overweight. Diabetes 25: 586–594, 1976.
25. Stamfer, MJ, Krauss, RM, Ma, J: A prospective study of triglyceride level, low density lipoprotein particle diameter, and risk of myocardial infarction. JAMA 276: 882–888, 1996.
26. Fontbonne, A, Thibault, N, Eschwege, E, Ducimetiere, P: Body fat distribution and coronary heart disease mortality in subjects with impaired glucose tolerance and diabetes mellitus: the Paris Prospective Study, 15-year follow-up. Diabetologia 35: 464–468, 1992.
27. Uusitupa, MIJ, Niskanen, LK, Sütonen, O, Voutilainen, E, Pyoröla, K: 5-year incidence of atheroscleotic vascular disease in relation to general risk factors, insulin level, and abnormalities in lipoprotein composition in non-insulin-dependent and non diabetic sub-jects. Circulation 82: 27–36, 1990.
28. Mitropoulos, KA: Hypercoagulability and factor VII in hypertriglyceridemia. Semin Thromb Hemost 14: 246–252, 1988.
29. Skartlien, AH, Lyberg-Beckmann, S, Holme, I, Hjermann, I, Pydz, H: Effect of alteration in triglyceride levels on factor VII-phospholipid complexes in plasma. Atherosclerosis 9: 798–80l, 1989.
30. Crutchley, DJ, McPhee, GV, Terris, MF, Canossa-Terris, MA: Levels of three hemostatic factors in relation to serum lipids: monocyte procoagulant activity, tissue plasminogen activator, and type-1 plasminogen activatir inhibitor. Atherosclerosis 9: 934–939, 1989.
31. Barrowclife, TW, Gray, E, Kerry, PJ, Gutteridge, JMC: Triglyceride-rich lipoproteins are responsible for thrombin generation induced by lipid peroxides. Thromb Haemost 52: 7–10, 1984.
32. Khoo, JC, Miller, E, McLoughlin, P, Steinberg, D: Prevention of low density lipoprotein aggregation by high density lipoprotein or apolipoprotein A-1. J Lipid Res 31: 645–658, 1990.
33. Parthasarathy, S, Barnett, J, Fong, LG: High density lipoprotein inhibits the oxidative modification of low density lipoprotein. Biochim Biophys Acta 1044: 275–283, 1990.
34. Romics L: Zsíranyagcsere-zavarok a mindennapi gyakorlatban. Golden Book, Budapest, 1995.
35. Romics L: Lipidanyagcsere-zavar és atherosclerosis diabetes mellitusban. [In: Halmos T, Jermendy Gy. (szerk.:) Diabetes mellitus. Medicina, Budapest, 1997] pp. 480–494.
36. Szollár L: A lipidanyagcsere zavarai. [In: Szollár L. (szerk.) Kórélettan, Semmelweis, Budapest, 1996] pp. 211–229.
37. Pollare, T, Lithell, H, Berne, C: Insulin resistance is a characteristic feature of primary hypertension independent of obesity. Metabolism 39: 167–174, 1990.
38. Bowie, A, Owens, D, Collins, P, Johnson, A, Tomkin, GH: Glycosylated low density lipoprotein is more sensitive to oxidation: implications for the diabetic patients? Atherosclerosis 102: 63–67. 1993.
39. Sobenin, IA, Tertov, VV, Koschinsky, T, Bunting, CE, Slavina, ES, Dedov, II, Orekhov, AN: Modified low-density lipoprotein from diabetic patients causes cholesterol accumulation in human intimal aortic cells. Atherosclerosis 100: 41 –54, 1993.
40. Lüscher, TF, Tanner, FC, Noll, G: Lipids and endothelial function: effects of lipid lowering and other therapeutic interventions. Curr Op Lipidol 7: 234–240, 1996.